Dedicated to spreading the Good News of Basic and Applied Science at great research institutions world wide. Good science is a collaborative process. The rule here: Science Never Sleeps.
Author: richardmitnick
68 years old, financial administrator, hiker, cyclist, PubRadio enthusiast
Gluons help to hold atoms together. (Sefa Kart/iStock/Getty Images Plus)
Scientists have long been on the lookout for ‘glueballs’, which are bound states of subatomic gluon particles on their own, without any quarks involved. Now, we may just have found them, hiding away in a particle accelerator experiment.
It promises to be a hugely significant breakthrough in physics, but for the benefit of everyone without a PhD in the subject, we’ll start at the beginning. The main job of gluons is to hold quarks in place and keep atoms stable – quarks being the building blocks that make up protons and neutrons.
This role makes the gluon part of the strong nuclear force – one of the four fundamental forces of nature that hold the laws of physics together, along with gravity, electromagnetism, and the weak nuclear force.
Until now, glueballs have only been theoretical propositions that physicists think should exist – because gluons should be able to stick to each other – rather than something that’s actually been observed.
Individual gluons don’t contain any matter, they just carry force, but glueballs do have mass created by the interactions of gluons. If we can spot them, it’s another indication that our current understanding of the way the Universe works, also known as the Standard Model of particle physics, is indeed right.
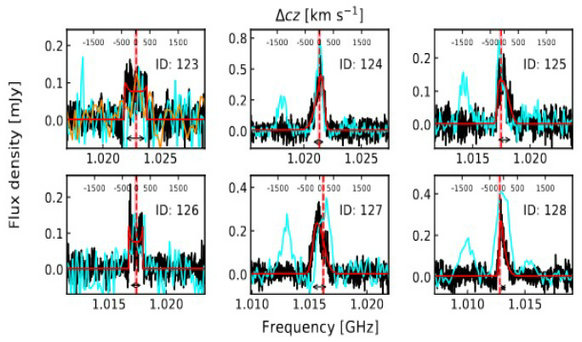
And so to the experiments at the Beijing Electron-Positron Collider II in China. The collider was used to smash together mesons, which are particles made up of a quark and antiquark held together by the strong nuclear force.
Sifting through the subatomic debris from these particle-smashing sessions – and we’re talking a decade of data involving some 10 billion samples – researchers were able to see evidence of particles with an average mass of 2,395 MeV/c^2. That’s the mass that glueballs are predicted to have.
The particle in question goes by the name X(2370), and while some of the other calculations involved don’t exactly fit what the researchers were looking for, they’re not far off. More measurements and more observations will be needed to get a definitive answer.
So it’s not quite proof of glueballs yet, but the evidence is starting to mount. Back in 2015, scientists also thought they had caught a glimpse of glueballs. Before too long, another particle might be making the jump from the theoretical to the actual.
A lot of this scientific research is made possible by continuing advances in mathematical techniques and computing capabilities – required to calculate the vast number of particular interactions and evolutions that are possible, and that might have originated from a glueball.
Plus of course, we now have the equipment and instruments necessary to peer into the most fundamental workings of the natural world, and to produce the billions of particle states required to spot something as rare and exotic as a glueball.
The Chinese Academy of Sciences[中国科学院](CN) is the national academy for the natural sciences of the People’s Republic of China. It has historical origins in the Academia Sinica during the Republican era and was formerly also known by that name. Collectively known as the “Two Academies (两院)” along with the Chinese Academy of Engineering, it functions as the national scientific think tank and academic governing body, providing advisory and appraisal services on issues stemming from the national economy, social development, and science and technology progress. It is headquartered in Xicheng District, Beijing, with branch institutes all over mainland China. It has also created hundreds of commercial enterprises, Lenovo being one of the most famous.
The Chinese Academy of Sciences has been ranked the No. 1 research institute in the world by Nature Index since the list’s inception in 2016 by Nature Portfolio. It is the most productive institution publishing articles of sustainable development indexed in Web of Science among all universities and research institutions in the world.
The Chinese Academy of Sciences has six academic divisions:
Chemistry (化学部)
Information Technological Sciences (信息技术科学部)
Earth Sciences (地学部)
Life Sciences and Medical Sciences (生命科学和医学学部)
Mathematics and Physics (数学物理学部)
Technological Sciences (技术科学部)
The CAS has thirteen regional branches, in Beijing, Shenyang, Changchun, Shanghai, Nanjing, Wuhan, Guangzhou, Chengdu, Kunming, Xi’an, Lanzhou, Hefei and Xinjiang. It has over one hundred institutes and four universities (the University of Science and Technology of China at Hefei, Anhui, the University of the Chinese Academy of Sciences in Beijing, ShanghaiTech University, and Shenzhen Institute of Advanced Technology). Backed by the institutes of CAS, UCAS is headquartered in Beijing, with graduate education bases in Shanghai, Chengdu, Wuhan, Guangzhou and Lanzhou, four Science Libraries of Chinese Academy of Sciences, three technology support centers and two news and publishing units. These CAS branches and offices are located in 20 provinces and municipalities throughout China. CAS has invested in or created over 430 science- and technology-based enterprises in eleven industries, including eight companies listed on stock exchanges.
Being granted a Fellowship of the Academy represents the highest level of national honor for Chinese scientists. The CAS membership system includes Academicians (院士), Emeritus Academicians (荣誉院士) and Foreign Academicians (外籍院士).
The Chinese Academy of Sciences was ranked very highly in the Nature Index Annual Tables, which measure the largest contributors to papers published in 82 leading journals.
Research institutes
Beijing Branch
University of the Chinese Academy of Sciences (UCAS)
Academy of Mathematics and Systems Science
Institute of Acoustics (IOA)
Institute of Atmospheric Physics
Institute of Botany, Chinese Academy of Sciences
Institute of Physics (IOPCAS)
Institute of Semiconductors
Institute of Electrical Engineering (IEE)
Institute of Information Engineering (IIE)
Institute of Theoretical Physics
Institute of High Energy Physics
Institute of Biophysics
Institute of Genetics and Developmental Biology
Institute of Electronics
National Astronomical Observatories
Institute of Computing Technology
Institute of Software
Institute of Automation
Beijing Institute of Genomics
Institute of Geographic Sciences and Natural Resources
Institute of Geology and Geophysics (IGG)
Institute of Remote Sensing and Digital Earth
Institute of Tibetan Plateau Research
Institute of Vertebrate Paleontology and Paleoanthropology
National Center for Nanoscience and Technology
Institute of Policy and Management
Institute of Psychology
Institute of Zoology
Changchun Branch
Changchun Institute of Optics, Fine Mechanics and Physics
Changchun Institute of Applied Chemistry
Northeast Institute of Geography and Agroecology
Changchun Observatory
Chengdu Branch
Institute of Mountain Hazards and Environment
Chengdu Institute of Biology
Institute of Optics and Electronics
Chengdu Institute of Organic Chemistry
Institute of Computer Application
Chongqing Institute of Green and Intelligent Technology
Guangzhou Branch
South China Botanical Garden
Shenzhen Institutes of Advanced Technology
South China Sea Institute of Oceanology
Guangzhou Institute of Energy Conversion
Guangzhou Institute of Geochemistry
Guangzhou Institute of Biomedicine and Health
Guiyang Branch
Institute of Geochemistry
Hefei Branch
Hefei Institutes of Physical Science
University of Science and Technology of China
Kunming Branch
Kunming Institute of Botany
Kunming Institute of Zoology
Xishuangbanna Tropical Botanical Garden
Institute of Geochemistry
Yunnan Astronomical Observatory
Lanzhou Branch
Institute of Modern Physics
Lanzhou Institute of Chemical Physics
Lanzhou Institute of Geology
Northwest Institute of Plateau Biology
Northwest Institute of Eco-Environment and Resources
Qinghai Institute of Salt Lakes Research
Nanjing Branch
Purple Mountain Observatory (Zijinshan Astronomical Observatory)
Institute of Soil Science
Nanjing Institute of Geology and Palaeontology
Nanjing Institute of Geography and Limnology
Nanjing Institute of Astronomical Optics and Technology
Suzhou Institute of Nano-tech and Nano-bionics (SINANO)
Suzhou Institute of Biomedical Engineering and Technology (SIBET)
Nanjing Botanical Garden, Memorial Sun Yat-Sen (Institute of Botany, Jiangsu Province and Chinese Academy of Science)
University of Chinese Academy of Sciences, Nanjing College
Shanghai Branch
Shanghai Astronomical Observatory
Shanghai Institute of Microsystem and Information Technology
Shanghai Institute of Technical Physics
Shanghai Institute of Optics and Fine Mechanics
Shanghai Institute of Ceramics
Shanghai Institute of Organic Chemistry
Shanghai Institute of Applied Physics
Shanghai Institutes for Biological Sciences
Shanghai Institute of Materia Medica
Institut Pasteur of Shanghai
Shanghai Advanced Research Institute, CAS
Institute of Neuroscience (ION)
ShanghaiTech University
Shenyang Branch
Institute of Metal Research
Shenyang Institute of Automation
Shenyang Institute of Applied Ecology, formerly the Institute of Forestry and Pedology
Shenyang Institute of Computing Technology
Dalian Institute of Chemical Physics
Qingdao Institute of Oceanology
Qingdao Institute of Bioenergy and Bioprocess Technology
Yantai Institute of Coastal Zone Research
Taiyuan Branch
Shanxi Institute of Coal Chemistry (ICCCAS)
Wuhan Branch
Wuhan Institute of Rock and Soil Mechanics
Wuhan Institute of Physics and Mathematics
Wuhan Institute of Virology
Institute of Geodesy and Geophysics
Institute of Hydrobiology
Wuhan Botanical Garden
Xinjiang Branch
Xinjiang Technical Institute of Physics and Chemistry
Xinjiang Institute of Ecology and Geography
Xi’an Branch
Xi’an Institute of Optics and Precision Mechanics
National Time Service Center
Institute of Earth Environment
In their ongoing quest to develop a range of methods for managing plasma so it can be used to generate electricity in a process known as fusion, researchers at the U.S. Department of Energy’s (DOE) Princeton Plasma Physics Laboratory (PPPL) have shown how two old methods can be combined to provide greater flexibility.
While the two methods – known as electron cyclotron current drive (“ECCD”) and applying resonant magnetic perturbations (RMP) – have long been studied, this is the first time researchers have simulated how they can be used together to give enhanced plasma control.
“This is kind of a new idea,” said Qiming Hu, a staff research physicist at PPPL and lead author of a new paper published in Nuclear Fusion about the work, which has also been demonstrated experimentally. “The full capabilities are still being figured out, but our paper does a great job of advancing our understanding of the potential benefits.”
Ultimately, scientists hope to use fusion to generate electricity. First, they will need to overcome several hurdles, including perfecting methods for minimizing bursts of particles from the plasma that are known as edge-localized modes (“ELMs”).
“Periodically, these bursts release a little bit of pressure because it’s too much. But these bursts can be dangerous,” said Hu, who works for PPPL at the DIII-D National Fusion Facility, a DOE user facility hosted by General Atomics.
ELMs can end a fusion reaction and even damage the tokamak, so researchers have developed many ways to try to avoid them.
“The best way we’ve found to avoid them is by applying resonant magnetic perturbations, or “RMPs”, that generate additional magnetic fields,” said PPPL Principal Research Physicist Alessandro Bortolon, who was one of the co-authors of the paper.
Magnetic fields generate islands, microwaves adjust them
The magnetic fields initially applied by the tokamak wind around the torus-shaped plasma, both the long way – around the outer edge, and the short way – from the outer edge and through the center hole. The additional magnetic fields created by the RMPs travel through the plasma, weaving in and out like a sewer’s stitch. These fields produce oval or circular magnetic fields in the plasma called magnetic islands.
“Normally, islands in plasmas are really, really bad. If the islands are too big, then the plasma itself can disrupt.”
However, the researchers already knew experimentally that under certain conditions, the islands can be beneficial. The hard part is generating RMPs big enough to generate the islands. That’s where the ECCD, which is basically a microwave beam injection, comes in. The researchers found that adding ECCD to the plasma’s edge lowers the amount of current required to generate the RMPs necessary to make the islands.
The image on the left shows the tokamak and 3D magnetic perturbation generated by 3D coils, with the purple-blue hues representing lower amplitude perturbations and the red representing higher amplitude perturbations. The image on the right is a closer view showing the top half of the tokamak and plasma. The coils are used to generate the magnetic field perturbations that produce the islands (blue). Another coil can also be found on the bottom of the machine. The injection system for the ECCD microwaves is depicted on top (red). These can be used to adjust the width of the islands. (Image credit: Qiming Hu / PPPL)
The microwave beam injection also allowed the researchers to perfect the size of the islands for maximum plasma edge stability. Metaphorically, the RMPs act like a simple light switch that turns the islands on, while the ECCD acts like an additional dimmer switch that lets the researchers adjust the islands to the ideal size for a manageable plasma.
“Our simulation refines our understanding of the interactions in play,” Hu said. “When the ECCD was added in the same direction as the current in the plasma, the width of the island decreased, and the pedestal pressure increased. Applying the ECCD in the opposite direction produced opposite results, with island width increasing and pedestal pressure dropping or facilitating island opening.”
ECCD at the edge, instead of the core
The research is also notable because ECCD was added to the plasma’s edge instead of the core, where it is typically used.
“Usually, people think applying localized ECCD at the plasma edge is risky because the microwaves may damage in-vessel components,” said Hu. “We’ve shown that it’s doable, and we’ve demonstrated the flexibility of the approach. This might open new avenues for designing future devices.”
By lowering the amount of current required to generate the RMPs, this simulation work could ultimately lead to lowering the cost of fusion energy production in commercial-scale fusion devices of the future.
This work was funded by the DOE under award numbers DE-AC02-09CH11466, DE-FC02-04ER54698, DE-SC0022270 and DE-AC52-07NA27344. Co-author Qingquan Yu was partially supported by the EUROfusion Enabling Research project (CfP-FSD-AWP24-ENR). His work has been carried out within the framework of the EUROfusion Consortium, funded by the European Union via the Euratom Research and Training Programme grant number 101052200.
The Princeton Plasma Physics Laboratory is a U.S. Department of Energy national laboratory managed by Princeton University. PPPL, on Princeton University’s Forrestal Campus in Plainsboro, N.J., is devoted to creating new knowledge about the physics of plasmas — ultra-hot, charged gases — and to developing practical solutions for the creation of fusion energy. Results of PPPL research have ranged from a portable nuclear materials detector for anti-terrorist use to universally employed computer codes for analyzing and predicting the outcome of fusion experiments. The Laboratory is managed by the University for the U.S. Department of Energy’s Office of Science, which is the largest single supporter of basic research in the physical sciences in the United States, and is working to address some of the most pressing challenges of our time. For more information, please visit https://energy.gov/science.
About Princeton: Overview
Princeton University is a private Ivy League research university in Princeton, New Jersey (US). Founded in 1746 in Elizabeth as the College of New Jersey, Princeton is the fourth-oldest institution of higher education in the United States and one of the nine colonial colleges chartered before the American Revolution. The institution moved to Newark in 1747, then to the current site nine years later. It was renamed Princeton University in 1896.
Princeton provides undergraduate and graduate instruction in the humanities, social sciences, natural sciences, and engineering. It offers professional degrees through the Princeton School of Public and International Affairs, the School of Engineering and Applied Science, the School of Architecture and the Bendheim Center for Finance. The university also manages the DOE’s Princeton Plasma Physics Laboratory. Princeton has the largest endowment per student in the United States.
As of October 2020, 69 Nobel laureates, 15 Fields Medalists and 14 Turing Award laureates have been affiliated with Princeton University as alumni, faculty members or researchers. In addition, Princeton has been associated with 21 National Medal of Science winners, 5 Abel Prize winners, 5 National Humanities Medal recipients, 215 Rhodes Scholars, 139 Gates Cambridge Scholars and 137 Marshall Scholars. Two U.S. Presidents, twelve U.S. Supreme Court Justices (three of whom currently serve on the court) and numerous living billionaires and foreign heads of state are all counted among Princeton’s alumni body. Princeton has also graduated many prominent members of the U.S. Congress and the U.S. Cabinet, including eight Secretaries of State, three Secretaries of Defense and the current Chairman of the Joint Chiefs of Staff.
Princeton University, founded as the College of New Jersey, was considered the successor of the “Log College” founded by the Reverend William Tennent at Neshaminy, PA in about 1726. New Light Presbyterians founded the College of New Jersey in 1746 in Elizabeth, New Jersey. Its purpose was to train ministers. The college was the educational and religious capital of Scottish Presbyterian America. Unlike Harvard University , which was originally “intensely English” with graduates taking the side of the crown during the American Revolution, Princeton was founded to meet the religious needs of the period and many of its graduates took the American side in the war. In 1754, trustees of the College of New Jersey suggested that, in recognition of Governor Jonathan Belcher’s interest, Princeton should be named as Belcher College. Belcher replied: “What a name that would be!” In 1756, the college moved its campus to Princeton, New Jersey. Its home in Princeton was Nassau Hall, named for the royal House of Orange-Nassau of William III of England.
Following the untimely deaths of Princeton’s first five presidents, John Witherspoon became president in 1768 and remained in that post until his death in 1794. During his presidency, Witherspoon shifted the college’s focus from training ministers to preparing a new generation for secular leadership in the new American nation. To this end, he tightened academic standards and solicited investment in the college. Witherspoon’s presidency constituted a long period of stability for the college, interrupted by the American Revolution and particularly the Battle of Princeton, during which British soldiers briefly occupied Nassau Hall; American forces, led by George Washington, fired cannon on the building to rout them from it.
In 1812, the eighth president of the College of New Jersey, Ashbel Green (1812–23), helped establish the Princeton Theological Seminary next door. The plan to extend the theological curriculum met with “enthusiastic approval on the part of the authorities at the College of New Jersey.” Today, Princeton University and Princeton Theological Seminary maintain separate institutions with ties that include services such as cross-registration and mutual library access.
Before the construction of Stanhope Hall in 1803, Nassau Hall was the college’s sole building. The cornerstone of the building was laid on September 17, 1754. During the summer of 1783, the Continental Congress met in Nassau Hall, making Princeton the country’s capital for four months. Over the centuries and through two redesigns following major fires (1802 and 1855), Nassau Hall’s role shifted from an all-purpose building, comprising office, dormitory, library, and classroom space; to classroom space exclusively; to its present role as the administrative center of the University. The class of 1879 donated twin lion sculptures that flanked the entrance until 1911, when that same class replaced them with tigers. Nassau Hall’s bell rang after the hall’s construction; however, the fire of 1802 melted it. The bell was then recast and melted again in the fire of 1855.
James McCosh became the college’s president in 1868 and lifted the institution out of a low period that had been brought about by the American Civil War. During his two decades of service, he overhauled the curriculum, oversaw an expansion of inquiry into the sciences, and supervised the addition of a number of buildings in the High Victorian Gothic style to the campus. McCosh Hall is named in his honor.
In 1879, the first thesis for a Doctor of Philosophy (Ph.D.) was submitted by James F. Williamson, Class of 1877.
In 1896, the college officially changed its name from the College of New Jersey to Princeton University to honor the town in which it resides. During this year, the college also underwent large expansion and officially became a university. In 1900, the Graduate School was established.
In 1902, Woodrow Wilson, graduate of the Class of 1879, was elected the 13th president of the university. Under Wilson, Princeton introduced the preceptorial system in 1905, a then-unique concept in the United States that augmented the standard lecture method of teaching with a more personal form in which small groups of students, or precepts, could interact with a single instructor, or preceptor, in their field of interest.
In 1906, the reservoir Carnegie Lake was created by Andrew Carnegie. A collection of historical photographs of the building of the lake is housed at the Seeley G. Mudd Manuscript Library on Princeton’s campus. On October 2, 1913, the Princeton University Graduate College was dedicated. In 1919 the School of Architecture was established. In 1933, Albert Einstein became a lifetime member of the Institute for Advanced Study with an office on the Princeton campus. While always independent of the university, the Institute for Advanced Study occupied offices in Jones Hall for 6 years, from its opening in 1933, until its own campus was finished and opened in 1939.
Coeducation
In 1969, Princeton University first admitted women as undergraduates. In 1887, the university actually maintained and staffed a sister college, Evelyn College for Women, in the town of Princeton on Evelyn and Nassau streets. It was closed after roughly a decade of operation. After abortive discussions with Sarah Lawrence College to relocate the women’s college to Princeton and merge it with the University in 1967, the administration decided to admit women and turned to the issue of transforming the school’s operations and facilities into a female-friendly campus. The administration had barely finished these plans in April 1969 when the admissions office began mailing out its acceptance letters. Its five-year coeducation plan provided $7.8 million for the development of new facilities that would eventually house and educate 650 women students at Princeton by 1974. Ultimately, 148 women, consisting of 100 freshmen and transfer students of other years, entered Princeton on September 6, 1969 amidst much media attention. Princeton enrolled its first female graduate student, Sabra Follett Meservey, as a PhD candidate in Turkish history in 1961. A handful of undergraduate women had studied at Princeton from 1963 on, spending their junior year there to study “critical languages” in which Princeton’s offerings surpassed those of their home institutions. They were considered regular students for their year on campus, but were not candidates for a Princeton degree.
As a result of a 1979 lawsuit by Sally Frank, Princeton’s eating clubs were required to go coeducational in 1991, after Tiger Inn’s appeal to the U.S. Supreme Court was denied. In 1987, the university changed the gendered lyrics of “Old Nassau” to reflect the school’s co-educational student body. From 2009 to 2011, Princeton professor Nannerl O. Keohane chaired a committee on undergraduate women’s leadership at the university, appointed by President Shirley M. Tilghman.
The main campus sits on about 500 acres (2.0 km^2) in Princeton. In 2011, the main campus was named by Travel+Leisure as one of the most beautiful in the United States. The James Forrestal Campus is split between nearby Plainsboro and South Brunswick. The University also owns some property in West Windsor Township. The campuses are situated about one hour from both New York City and Philadelphia.
The first building on campus was Nassau Hall, completed in 1756 and situated on the northern edge of campus facing Nassau Street. The campus expanded steadily around Nassau Hall during the early and middle 19th century. The McCosh presidency (1868–88) saw the construction of a number of buildings in the High Victorian Gothic and Romanesque Revival styles; many of them are now gone, leaving the remaining few to appear out of place. At the end of the 19th century much of Princeton’s architecture was designed by the Cope and Stewardson firm (same architects who designed a large part of Washington University in St Louis and University of Pennsylvania) resulting in the Collegiate Gothic style for which it is known today. Implemented initially by William Appleton Potter and later enforced by the University’s supervising architect, Ralph Adams Cram, the Collegiate Gothic style remained the standard for all new building on the Princeton campus through 1960. A flurry of construction in the 1960s produced a number of new buildings on the south side of the main campus, many of which have been poorly received. Several prominent architects have contributed some more recent additions, including Frank Gehry (Lewis Library), I. M. Pei (Spelman Halls), Demetri Porphyrios (Whitman College, a Collegiate Gothic project), Robert Venturi and Denise Scott Brown (Frist Campus Center, among several others), and Rafael Viñoly (Carl Icahn Laboratory).
A group of 20th-century sculptures scattered throughout the campus forms the Putnam Collection of Sculpture. It includes works by Alexander Calder (Five Disks: One Empty), Jacob Epstein (Albert Einstein), Henry Moore (Oval with Points), Isamu Noguchi (White Sun), and Pablo Picasso (Head of a Woman). Richard Serra’s The Hedgehog and The Fox is located between Peyton and Fine halls next to Princeton Stadium and the Lewis Library.
At the southern edge of the campus is Carnegie Lake, an artificial lake named for Andrew Carnegie. Carnegie financed the lake’s construction in 1906 at the behest of a friend who was a Princeton alumnus. Carnegie hoped the opportunity to take up rowing would inspire Princeton students to forsake football, which he considered “not gentlemanly.” The Shea Rowing Center on the lake’s shore continues to serve as the headquarters for Princeton rowing.
Cannon Green
Buried in the ground at the center of the lawn south of Nassau Hall is the “Big Cannon,” which was left in Princeton by British troops as they fled following the Battle of Princeton. It remained in Princeton until the War of 1812, when it was taken to New Brunswick. In 1836 the cannon was returned to Princeton and placed at the eastern end of town. It was removed to the campus under cover of night by Princeton students in 1838 and buried in its current location in 1840.
A second “Little Cannon” is buried in the lawn in front of nearby Whig Hall. This cannon, which may also have been captured in the Battle of Princeton, was stolen by students of Rutgers University in 1875. The theft ignited the Rutgers-Princeton Cannon War. A compromise between the presidents of Princeton and Rutgers ended the war and forced the return of the Little Cannon to Princeton. The protruding cannons are occasionally painted scarlet by Rutgers students who continue the traditional dispute.
In years when the Princeton football team beats the teams of both Harvard University and Yale University in the same season, Princeton celebrates with a bonfire on Cannon Green. This occurred in 2012, ending a five-year drought. The next bonfire happened on November 24, 2013, and was broadcast live over the Internet.
Landscape
Princeton’s grounds were designed by Beatrix Farrand between 1912 and 1943. Her contributions were most recently recognized with the naming of a courtyard for her. Subsequent changes to the landscape were introduced by Quennell Rothschild & Partners in 2000. In 2005, Michael Van Valkenburgh was hired as the new consulting landscape architect for the campus. Lynden B. Miller was invited to work with him as Princeton’s consulting gardening architect, focusing on the 17 gardens that are distributed throughout the campus.
Buildings
Nassau Hall
Nassau Hall is the oldest building on campus. Begun in 1754 and completed in 1756, it was the first seat of the New Jersey Legislature in 1776, was involved in the battle of Princeton in 1777, and was the seat of the Congress of the Confederation (and thus capitol of the United States) from June 30, 1783, to November 4, 1783. It now houses the office of the university president and other administrative offices, and remains the symbolic center of the campus. The front entrance is flanked by two bronze tigers, a gift of the Princeton Class of 1879. Commencement is held on the front lawn of Nassau Hall in good weather. In 1966, Nassau Hall was added to the National Register of Historic Places.
Residential colleges
Princeton has six undergraduate residential colleges, each housing approximately 500 freshmen, sophomores, some juniors and seniors, and a handful of junior and senior resident advisers. Each college consists of a set of dormitories, a dining hall, a variety of other amenities—such as study spaces, libraries, performance spaces, and darkrooms—and a collection of administrators and associated faculty. Two colleges, First College and Forbes College (formerly Woodrow Wilson College and Princeton Inn College, respectively), date to the 1970s; three others, Rockefeller, Mathey, and Butler Colleges, were created in 1983 following the Committee on Undergraduate Residential Life (CURL) report, which suggested the institution of residential colleges as a solution to an allegedly fragmented campus social life. The construction of Whitman College, the university’s sixth residential college, was completed in 2007.
Rockefeller and Mathey are located in the northwest corner of the campus; Princeton brochures often feature their Collegiate Gothic architecture. Like most of Princeton’s Gothic buildings, they predate the residential college system and were fashioned into colleges from individual dormitories.
First and Butler, located south of the center of the campus, were built in the 1960s. First served as an early experiment in the establishment of the residential college system. Butler, like Rockefeller and Mathey, consisted of a collection of ordinary dorms (called the “New New Quad”) before the addition of a dining hall made it a residential college. Widely disliked for their edgy modernist design, including “waffle ceilings,” the dormitories on the Butler Quad were demolished in 2007. Butler is now reopened as a four-year residential college, housing both under- and upperclassmen.
Forbes is located on the site of the historic Princeton Inn, a gracious hotel overlooking the Princeton golf course. The Princeton Inn, originally constructed in 1924, played regular host to important symposia and gatherings of renowned scholars from both the university and the nearby Institute for Advanced Study for many years. Forbes currently houses nearly 500 undergraduates in its residential halls.
In 2003, Princeton broke ground for a sixth college named Whitman College after its principal sponsor, Meg Whitman, who graduated from Princeton in 1977. The new dormitories were constructed in the Collegiate Gothic architectural style and were designed by architect Demetri Porphyrios. Construction finished in 2007, and Whitman College was inaugurated as Princeton’s sixth residential college that same year.
The precursor of the present college system in America was originally proposed by university president Woodrow Wilson in the early 20th century. For over 800 years, however, the collegiate system had already existed in Britain at University of Cambridge (UK) and University of Oxford (UK). Wilson’s model was much closer to Yale University’s present system, which features four-year colleges. Lacking the support of the trustees, the plan languished until 1968. That year, Wilson College was established to cap a series of alternatives to the eating clubs. Fierce debates raged before the present residential college system emerged. The plan was first attempted at Yale, but the administration was initially uninterested; an exasperated alumnus, Edward Harkness, finally paid to have the college system implemented at Harvard in the 1920s, leading to the oft-quoted aphorism that the college system is a Princeton idea that was executed at Harvard with funding from Yale.
Princeton has one graduate residential college, known simply as the Graduate College, located beyond Forbes College at the outskirts of campus. The far-flung location of the GC was the spoil of a squabble between Woodrow Wilson and then-Graduate School Dean Andrew Fleming West. Wilson preferred a central location for the college; West wanted the graduate students as far as possible from the campus. Ultimately, West prevailed. The Graduate College is composed of a large Collegiate Gothic section crowned by Cleveland Tower, a local landmark that also houses a world-class carillon. The attached New Graduate College provides a modern contrast in architectural style.
McCarter Theatre
The Tony-award-winning McCarter Theatre was built by the Princeton Triangle Club, a student performance group, using club profits and a gift from Princeton University alumnus Thomas McCarter. Today, the Triangle Club performs its annual freshmen revue, fall show, and Reunions performances in McCarter. McCarter is also recognized as one of the leading regional theaters in the United States.
Art Museum
The Princeton University Art Museum was established in 1882 to give students direct, intimate, and sustained access to original works of art that complement and enrich instruction and research at the university. This continues to be a primary function, along with serving as a community resource and a destination for national and international visitors.
Numbering over 92,000 objects, the collections range from ancient to contemporary art and concentrate geographically on the Mediterranean regions, Western Europe, China, the United States, and Latin America. There is a collection of Greek and Roman antiquities, including ceramics, marbles, bronzes, and Roman mosaics from faculty excavations in Antioch. Medieval Europe is represented by sculpture, metalwork, and stained glass. The collection of Western European paintings includes examples from the early Renaissance through the 19th century, with masterpieces by Monet, Cézanne, and Van Gogh, and features a growing collection of 20th-century and contemporary art, including iconic paintings such as Andy Warhol’s Blue Marilyn.
One of the best features of the museums is its collection of Chinese art, with important holdings in bronzes, tomb figurines, painting, and calligraphy. Its collection of pre-Columbian art includes examples of Mayan art, and is commonly considered to be the most important collection of pre-Columbian art outside of Latin America. The museum has collections of old master prints and drawings and a comprehensive collection of over 27,000 original photographs. African art and Northwest Coast Indian art are also represented. The Museum also oversees the outdoor Putnam Collection of Sculpture.
University Chapel
The Princeton University Chapel is located on the north side of campus, near Nassau Street. It was built between 1924 and 1928, at a cost of $2.3 million [approximately $34.2 million in 2020 dollars]. Ralph Adams Cram, the University’s supervising architect, designed the chapel, which he viewed as the crown jewel for the Collegiate Gothic motif he had championed for the campus. At the time of its construction, it was the second largest university chapel in the world, after King’s College Chapel, Cambridge. It underwent a two-year, $10 million restoration campaign between 2000 and 2002.
Measured on the exterior, the chapel is 277 feet (84 m) long, 76 feet (23 m) wide at its transepts, and 121 feet (37 m) high. The exterior is Pennsylvania sandstone, with Indiana limestone used for the trim. The interior is mostly limestone and Aquia Creek sandstone. The design evokes an English church of the Middle Ages. The extensive iconography, in stained glass, stonework, and wood carvings, has the common theme of connecting religion and scholarship.
The Chapel seats almost 2,000. It hosts weekly ecumenical Christian services, daily Roman Catholic mass, and several annual special events.
Murray-Dodge Hall
Murray-Dodge Hall houses the Office of Religious Life (ORL), the Murray Dodge Theater, the Murray-Dodge Café, the Muslim Prayer Room and the Interfaith Prayer Room. The ORL houses the office of the Dean of Religious Life, Alison Boden, and a number of university chaplains, including the country’s first Hindu chaplain, Vineet Chander; and one of the country’s first Muslim chaplains, Sohaib Sultan.
Sustainability
Published in 2008, Princeton’s Sustainability Plan highlights three priority areas for the University’s Office of Sustainability: reduction of greenhouse gas emissions; conservation of resources; and research, education, and civic engagement. Princeton has committed to reducing its carbon dioxide emissions to 1990 levels by 2020: Energy without the purchase of offsets. The University published its first Sustainability Progress Report in November 2009. The University has adopted a green purchasing policy and recycling program that focuses on paper products, construction materials, lightbulbs, furniture, and electronics. Its dining halls have set a goal to purchase 75% sustainable food products by 2015. The student organization “Greening Princeton” seeks to encourage the University administration to adopt environmentally friendly policies on campus.
Organization
The Trustees of Princeton University, a 40-member board, is responsible for the overall direction of the University. It approves the operating and capital budgets, supervises the investment of the University’s endowment and oversees campus real estate and long-range physical planning. The trustees also exercise prior review and approval concerning changes in major policies, such as those in instructional programs and admission, as well as tuition and fees and the hiring of faculty members.
With an endowment of $26.1 billion, Princeton University is among the wealthiest universities in the world. Ranked in 2010 as the third largest endowment in the United States, the university had the greatest per-student endowment in the world (over $2 million for undergraduates) in 2011. Such a significant endowment is sustained through the continued donations of its alumni and is maintained by investment advisers. Some of Princeton’s wealth is invested in its art museum, which features works by Claude Monet, Vincent van Gogh, Jackson Pollock, and Andy Warhol among other prominent artists.
Academics
Undergraduates fulfill general education requirements, choose among a wide variety of elective courses, and pursue departmental concentrations and interdisciplinary certificate programs. Required independent work is a hallmark of undergraduate education at Princeton. Students graduate with either the Bachelor of Arts (A.B.) or the Bachelor of Science in Engineering (B.S.E.).
The graduate school offers advanced degrees spanning the humanities, social sciences, natural sciences, and engineering. Doctoral education is available in most disciplines. It emphasizes original and independent scholarship whereas master’s degree programs in architecture, engineering, finance, and public affairs and public policy prepare candidates for careers in public life and professional practice.
Undergraduate courses in the humanities are traditionally either seminars or lectures held 2 or 3 times a week with an additional discussion seminar that is called a “precept.” To graduate, all A.B. candidates must complete a senior thesis and, in most departments, one or two extensive pieces of independent research that are known as “junior papers.” Juniors in some departments, including architecture and the creative arts, complete independent projects that differ from written research papers. A.B. candidates must also fulfill a three or four semester foreign language requirement and distribution requirements (which include, for example, classes in ethics, literature and the arts, and historical analysis) with a total of 31 classes. B.S.E. candidates follow a parallel track with an emphasis on a rigorous science and math curriculum, a computer science requirement, and at least two semesters of independent research including an optional senior thesis. All B.S.E. students must complete at least 36 classes. A.B. candidates typically have more freedom in course selection than B.S.E. candidates because of the fewer number of required classes. Nonetheless, in the spirit of a liberal arts education, both enjoy a comparatively high degree of latitude in creating a self-structured curriculum.
Undergraduates agree to adhere to an academic integrity policy called the Honor Code, established in 1893. Under the Honor Code, faculty do not proctor examinations; instead, the students proctor one another and must report any suspected violation to an Honor Committee made up of undergraduates. The Committee investigates reported violations and holds a hearing if it is warranted. An acquittal at such a hearing results in the destruction of all records of the hearing; a conviction results in the student’s suspension or expulsion. The signed pledge required by the Honor Code is so integral to students’ academic experience that the Princeton Triangle Club performs a song about it each fall. Out-of-class exercises fall under the jurisdiction of the Faculty-Student Committee on Discipline. Undergraduates are expected to sign a pledge on their written work affirming that they have not plagiarized the work.
Graduate
The Graduate School has about 2,600 students in 42 academic departments and programs in social sciences; engineering; natural sciences; and humanities. These departments include the Department of Psychology; Department of History; and Department of Economics.
In 2017–2018, it received nearly 11,000 applications for admission and accepted around 1,000 applicants. The University also awarded 319 Ph.D. degrees and 170 final master’s degrees. Princeton has no medical school, law school, business school, or school of education. (A short-lived Princeton Law School folded in 1852.) It offers professional graduate degrees in architecture; engineering; finance and public policy- the last through the Princeton School of Public and International Affairs founded in 1930 as the School of Public and International Affairs and renamed in 1948 after university president (and U.S. president) Woodrow Wilson, and most recently renamed in 2020.
Libraries
The Princeton University Library system houses over eleven million holdings including seven million bound volumes. The main university library, Firestone Library, which houses almost four million volumes, is one of the largest university libraries in the world. Additionally, it is among the largest “open stack” libraries in existence. Its collections include the autographed manuscript of F. Scott Fitzgerald’s The Great Gatsby and George F. Kennan’s Long Telegram. In addition to Firestone library, specialized libraries exist for architecture, art and archaeology, East Asian studies, engineering, music, public and international affairs, public policy and university archives, and the sciences. In an effort to expand access, these libraries also subscribe to thousands of electronic resources.
Institutes
High Meadows Environmental Institute
The High Meadows Environmental Institute is an “interdisciplinary center of environmental research, education, and outreach” at the university. The institute was started in 1994. About 90 faculty members at Princeton University are affiliated with it.
The High Meadows Environmental Institute has the following research centers:
Carbon Mitigation Initiative (CMI): This is a 15-year-long partnership between PEI and British Petroleum with the goal of finding solutions to problems related to climate change. The Stabilization Wedge Game has been created as part of this initiative.
Center for BioComplexity (CBC)
Cooperative Institute for Climate Science (CICS): This is a collaboration with the National Oceanographic and Atmospheric Administration’s Geophysical Fluid Dynamics Laboratory.
Energy Systems Analysis Group
Grand Challenges
The DOE’s Princeton Plasma Physics Laboratory was founded in 1951 as Project Matterhorn, a top-secret cold war project aimed at achieving controlled nuclear fusion. Princeton astrophysics professor Lyman Spitzer became the first director of the project and remained director until the lab’s declassification in 1961 when it received its current name.
PPPL currently houses approximately half of the graduate astrophysics department, the Princeton Program in Plasma Physics. The lab is also home to the Harold P. Furth Plasma Physics Library. The library contains all declassified Project Matterhorn documents, included the first design sketch of a stellarator by Lyman Spitzer.
Princeton is one of five US universities to have and to operate a Department of Energy national laboratory.
Student life and culture
University housing is guaranteed to all undergraduates for all four years. More than 98% of students live on campus in dormitories. Freshmen and sophomores must live in residential colleges, while juniors and seniors typically live in designated upperclassman dormitories. The actual dormitories are comparable, but only residential colleges have dining halls. Nonetheless, any undergraduate may purchase a meal plan and eat in a residential college dining hall. Recently, upperclassmen have been given the option of remaining in their college for all four years. Juniors and seniors also have the option of living off-campus, but high rent in the Princeton area encourages almost all students to live in university housing. Undergraduate social life revolves around the residential colleges and a number of coeducational eating clubs, which students may choose to join in the spring of their sophomore year. Eating clubs, which are not officially affiliated with the university, serve as dining halls and communal spaces for their members and also host social events throughout the academic year.
Princeton’s six residential colleges host a variety of social events and activities, guest speakers, and trips. The residential colleges also sponsor trips to New York for undergraduates to see ballets, operas, Broadway shows, sports events, and other activities. The eating clubs, located on Prospect Avenue, are co-ed organizations for upperclassmen. Most upperclassmen eat their meals at one of the eleven eating clubs. Additionally, the clubs serve as evening and weekend social venues for members and guests. The eleven clubs are Cannon; Cap and Gown; Charter; Cloister; Colonial; Cottage; Ivy; Quadrangle; Terrace; Tiger; and Tower.
Princeton hosts two Model United Nations conferences, PMUNC in the fall for high school students and PDI in the spring for college students. It also hosts the Princeton Invitational Speech and Debate tournament each year at the end of November. Princeton also runs Princeton Model Congress, an event that is held once a year in mid-November. The four-day conference has high school students from around the country as participants.
Although the school’s admissions policy is need-blind, Princeton, based on the proportion of students who receive Pell Grants, was ranked as a school with little economic diversity among all national universities ranked by U.S. News & World Report. While Pell figures are widely used as a gauge of the number of low-income undergraduates on a given campus, the rankings article cautions “the proportion of students on Pell Grants isn’t a perfect measure of an institution’s efforts to achieve economic diversity,” but goes on to say that “still, many experts say that Pell figures are the best available gauge of how many low-income undergrads there are on a given campus.”
TigerTrends is a university-based student run fashion, arts, and lifestyle magazine.
Demographics
Princeton has made significant progress in expanding the diversity of its student body in recent years. The 2019 freshman class was one of the most diverse in the school’s history, with 61% of students identifying as students of color. Undergraduate and master’s students were 51% male and 49% female for the 2018–19 academic year.
The median family income of Princeton students is $186,100, with 57% of students coming from the top 10% highest-earning families and 14% from the bottom 60%.
In 1999, 10% of the student body was Jewish, a percentage lower than those at other Ivy League schools. Sixteen percent of the student body was Jewish in 1985; the number decreased by 40% from 1985 to 1999. This decline prompted The Daily Princetonian to write a series of articles on the decline and its reasons. Caroline C. Pam of The New York Observer wrote that Princeton was “long dogged by a reputation for anti-Semitism” and that this history as well as Princeton’s elite status caused the university and its community to feel sensitivity towards the decrease of Jewish students. At the time many Jewish students at Princeton dated Jewish students at the University of Pennsylvania in Philadelphia because they perceived Princeton as an environment where it was difficult to find romantic prospects; Pam stated that there was a theory that the dating issues were a cause of the decline in Jewish students.
In 1981, the population of African Americans at Princeton University made up less than 10%. Bruce M. Wright was admitted into the university in 1936 as the first African American, however, his admission was a mistake and when he got to campus he was asked to leave. Three years later Wright asked the dean for an explanation on his dismissal and the dean suggested to him that “a member of your race might feel very much alone” at Princeton University.
Traditions
Princeton enjoys a wide variety of campus traditions, some of which, like the Clapper Theft and Nude Olympics, have faded into history:
Arch Sings – Late-night concerts that feature one or several of Princeton’s undergraduate a cappella groups, such as the Princeton Nassoons; Princeton Tigertones; Princeton Footnotes; Princeton Roaring 20; and The Princeton Wildcats. The free concerts take place in one of the larger arches on campus. Most are held in Blair Arch or Class of 1879 Arch.
Bonfire – Ceremonial bonfire that takes place in Cannon Green behind Nassau Hall. It is held only if Princeton beats both Harvard University and Yale University at football in the same season. The most recent bonfire was lighted on November 18, 2018.
Bicker – Selection process for new members that is employed by selective eating clubs. Prospective members, or bickerees, are required to perform a variety of activities at the request of current members.
Cane Spree – An athletic competition between freshmen and sophomores that is held in the fall. The event centers on cane wrestling, where a freshman and a sophomore will grapple for control of a cane. This commemorates a time in the 1870s when sophomores, angry with the freshmen who strutted around with fancy canes, stole all of the canes from the freshmen, hitting them with their own canes in the process.
The Clapper or Clapper Theft – The act of climbing to the top of Nassau Hall to steal the bell clapper, which rings to signal the start of classes on the first day of the school year. For safety reasons, the clapper has been removed permanently.
Class Jackets (Beer Jackets) – Each graduating class designs a Class Jacket that features its class year. The artwork is almost invariably dominated by the school colors and tiger motifs.
Communiversity – An annual street fair with performances, arts and crafts, and other activities that attempts to foster interaction between the university community and the residents of Princeton.
Dean’s Date – The Tuesday at the end of each semester when all written work is due. This day signals the end of reading period and the beginning of final examinations. Traditionally, undergraduates gather outside McCosh Hall before the 5:00 PM deadline to cheer on fellow students who have left their work to the very last minute.
FitzRandolph Gates – At the end of Princeton’s graduation ceremony, the new graduates process out through the main gate of the university as a symbol of the fact that they are leaving college. According to tradition, anyone who exits campus through the FitzRandolph Gates before his or her own graduation date will not graduate.
Holder Howl – The midnight before Dean’s Date, students from Holder Hall and elsewhere gather in the Holder courtyard and take part in a minute-long, communal primal scream to vent frustration from studying with impromptu, late night noise making.
Houseparties – Formal parties that are held simultaneously by all of the eating clubs at the end of the spring term.
Ivy stones – Class memorial stones placed on the exterior walls of academic buildings around the campus.
Lawnparties – Parties that feature live bands that are held simultaneously by all of the eating clubs at the start of classes and at the conclusion of the academic year.
Princeton Locomotive – Traditional cheer in use since the 1890s. It is commonly heard at Opening Exercises in the fall as alumni and current students welcome the freshman class, as well as the P-rade in the spring at Princeton Reunions. The cheer starts slowly and picks up speed, and includes the sounds heard at a fireworks show.
Or if a class is being celebrated, the last line consists of the class year repeated three times, e.g. “Eighty-eight! Eighty-eight! Eighty-eight!”
Newman’s Day – Students attempt to drink 24 beers in the 24 hours of April 24. According to The New York Times, “the day got its name from an apocryphal quote attributed to Paul Newman: ’24 beers in a case, 24 hours in a day. Coincidence? I think not.'” Newman had spoken out against the tradition, however.
Nude Olympics – Annual nude and partially nude frolic in Holder Courtyard that takes place during the first snow of the winter. Started in the early 1970s, the Nude Olympics went co-educational in 1979 and gained much notoriety with the American press. For safety reasons, the administration banned the Olympics in 2000 to the chagrin of students.
Prospect 11 – The act of drinking a beer at all 11 eating clubs in a single night.
P-rade – Traditional parade of alumni and their families. They process through campus by class year during Reunions.
Reunions – Massive annual gathering of alumni held the weekend before graduation.
Athletics
Princeton supports organized athletics at three levels: varsity intercollegiate, club intercollegiate, and intramural. It also provides “a variety of physical education and recreational programs” for members of the Princeton community. According to the athletics program’s mission statement, Princeton aims for its students who participate in athletics to be “‘student athletes’ in the fullest sense of the phrase. Most undergraduates participate in athletics at some level.
Princeton’s colors are orange and black. The school’s athletes are known as Tigers, and the mascot is a tiger. The Princeton administration considered naming the mascot in 2007, but the effort was dropped in the face of alumni opposition.
Varsity
Princeton is an NCAA Division I school. Its athletic conference is the Ivy League. Princeton hosts 38 men’s and women’s varsity sports. The largest varsity sport is rowing, with almost 150 athletes.
Princeton’s football team has a long and storied history. Princeton played against Rutgers University in the first intercollegiate football game in the U.S. on Nov 6, 1869. By a score of 6–4, Rutgers won the game, which was played by rules similar to modern rugby. Today Princeton is a member of the Football Championship Subdivision of NCAA Division I. As of the end of the 2010 season, Princeton had won 26 national football championships, more than any other school.
Club and intramural
In addition to varsity sports, Princeton hosts about 35 club sports teams. Princeton’s rugby team is organized as a club sport. Princeton’s sailing team is also a club sport, though it competes at the varsity level in the MAISA conference of the Inter-Collegiate Sailing Association.
Each year, nearly 300 teams participate in intramural sports at Princeton. Intramurals are open to members of Princeton’s faculty, staff, and students, though a team representing a residential college or eating club must consist only of members of that college or club. Several leagues with differing levels of competitiveness are available.
Songs
Notable among a number of songs commonly played and sung at various events such as commencement, convocation, and athletic games is Princeton Cannon Song, the Princeton University fight song.
Bob Dylan wrote Day of The Locusts (for his 1970 album New Morning) about his experience of receiving an honorary doctorate from the University. It is a reference to the negative experience he had and it mentions the Brood X cicada infestation Princeton experienced that June 1970.
“Old Nassau”
Old Nassau has been Princeton University’s anthem since 1859. Its words were written that year by a freshman, Harlan Page Peck, and published in the March issue of the Nassau Literary Review (the oldest student publication at Princeton and also the second oldest undergraduate literary magazine in the country). The words and music appeared together for the first time in Songs of Old Nassau, published in April 1859. Before the Langlotz tune was written, the song was sung to Auld Lang Syne’s melody, which also fits.
However, Old Nassau does not only refer to the university’s anthem. It can also refer to Nassau Hall, the building that was built in 1756 and named after William III of the House of Orange-Nassau. When built, it was the largest college building in North America. It served briefly as the capitol of the United States when the Continental Congress convened there in the summer of 1783. By metonymy, the term can refer to the university as a whole. Finally, it can also refer to a chemical reaction that is dubbed “Old Nassau reaction” because the solution turns orange and then black.
New research into molten rock 20km below the Earth’s surface could help save lives by improving the prediction of volcanic activity.
Volcanic eruptions pose significant hazards, with devastating impacts on both people living nearby and the environment.
They are currently predicted based on activity of the volcano itself and the upper few kilometres of crust beneath it, which contains molten rock potentially ready to erupt.
However, new research highlights the importance of searching for clues much deeper in the Earth’s crust, where rocks are first melted into magma before rising to chambers closer to the surface.
To understand the inner workings of our planet’s most explosive phenomena, researchers at Imperial College London and the University of Bristol dug deep to shed light on the frequency, composition, and size of volcanic eruptions around the world.
Their findings suggest that the size and frequency of eruptions are closely linked to the time it takes for extremely hot, molten rock known as magma to form in these deep reservoirs beneath the Earth’s crust – at depths of up to 20 kilometres – as well as to the size of these reservoirs.
Researchers believe that the findings, published in Science Advances, will allow them to predict volcanic eruptions more accurately, ultimately safeguarding communities of people and helping mitigate risks to the environment.
Studying volcanoes around the world
The study, led by researchers at the Department of Earth Science and Engineering at Imperial, reviewed data from 60 of the most explosive volcanic eruptions, spanning nine countries: the United States, New Zealand, Japan, Russia, Argentina, Chile, Nicaragua, El Salvador and Indonesia.
Study author Dr Catherine Booth, Research Associate in the Department of Earth Science and Engineering at Imperial College London, said:
“We looked at volcanoes around the world and dug deeper than previous studies that focused on shallow underground chambers where magma is stored before eruptions. We focused on understanding magma source reservoirs deep beneath our feet, where extreme heat melts solid rocks into magma at depths of around 10 to 20 kilometres.”
The team combined real-world data with advanced computer models. They looked at the composition, structure, and history of rocks deep beneath the Earth’s crust, alongside information gathered from active volcanoes, to understand how magma builds up and behaves deep underground, eventually rising through the Earth’s crust to volcanoes.
Using this information, researchers created computer simulations that mimic the complex processes of magma flow and storage deep within the Earth. Through these simulations, the team gained new insights into what factors drive volcanic eruptions.
Identifying key controls of eruptions
“Contrary to previous beliefs, our study suggests that the buoyancy of the magma, rather than the proportion of solid and molten rock, is what drives eruptions,” said Dr Booth.
“Magma buoyancy is controlled by its temperature and chemical composition compared to the surrounding rock– as the magma accumulates its composition changes to make it less dense, making it more ‘buoyant’ and enabling it to rise.
“Once the magma becomes buoyant enough to float, it rises and creates fractures in the overlying solid rock – and it then flows through these fractures very rapidly, causing an eruption.”
As well as identifying buoyancy of magma as an important factor driving eruptions, researchers also looked at how magma behaves once it reaches shallower underground chambers right before erupting. They found that how long magma was stored in these shallower chambers can have an effect on volcanic eruptions too – with longer periods of storage leading to smaller eruptions.
While larger reservoirs may be expected to fuel greater, more explosive eruptions, the findings also revealed that very large reservoirs disperse heat, which slows down the process of melting solid rocks into magma. This led researchers to conclude that the size of reservoirs is another key factor for predicting eruption sizes accurately – and that there is such a thing as an optimal size for the most explosive eruptions.
Findings also highlight that eruptions are rarely isolated and, instead, are part of a repetitive cycle. Additionally, the magma released by the volcanoes they studied was high in silica, a natural compound known to play a role in determining the viscosity and explosiveness of magma – with high-silica magma tending to be more viscous and resulting in more explosive eruptions.
Next steps
Co-author Professor Matt Jackson, Chair in Geological Fluid Dynamics in the Department of Earth Science and Engineering at Imperial College London, said:
“By improving our understanding of the processes behind volcanic activity and providing models that shed light on the factors controlling eruptions, our study is a crucial step towards better monitoring and forecasting of these powerful geological events.
“Our study had some limitations: our model focused on how magma flows upwards, and the source reservoirs in our model contained only molten rock and crystals. However, there is evidence that other fluids such as water and carbon dioxide are also found in these source reservoirs, and that magma can swirl and flow sideways.”
The next steps for researchers will be to refine their models, incorporating three-dimensional flow and accounting for different fluid compositions. In this way, they hope to continue to decipher the Earth’s processes responsible for volcanic eruptions – helping us better prepare for natural disasters in the future.
Imperial College London (UK) is a science-based university with an international reputation for excellence in teaching and research. Consistently rated amongst the world’s best universities, Imperial is committed to developing the next generation of researchers, scientists and academics through collaboration across disciplines. Located in the heart of London, Imperial is a multidisciplinary space for education, research, translation and commercialization, harnessing science and innovation to tackle global challenges.
Imperial College London (legally Imperial College of Science, Technology and Medicine) is a public research university in London. Imperial grew out of Prince Albert’s vision of an area for culture, including the Royal Albert Hall; Imperial Institute; numerous museums and the Royal Colleges that would go on to form the college. In 1907, Imperial College was established by Royal Charter, merging the Royal College of Science; Royal School of Mines; and City and Guilds College. In 1988, the Imperial College School of Medicine was formed by combining with St Mary’s Hospital Medical School. In 2004, Queen Elizabeth II opened the Imperial College Business School.
The college focuses exclusively on science; technology; medicine; and business. The college’s main campus is located in South Kensington, and it has an innovation campus in White City; a research field station at Silwood Park; and teaching hospitals throughout London. The college was a member of the University of London (UK) from 1908, becoming independent on its centenary in 2007. Imperial has an international community, with more than 59% of students from outside the UK and 140 countries represented on campus. Student, staff, and researcher affiliations include Nobel laureates, Fields Medalists, Breakthrough Prize winners, Turing Award winners; Fellows of the Royal Society, Fellows of the Royal Academy of Engineering and Fellows of the Academy of Medical Sciences.
History
19th century
The earliest college that led to the formation of Imperial was the Royal College of Chemistry founded in 1845 with the support of Prince Albert and parliament. This was merged in 1853 into what became known as the Royal School of Mines. The medical school has roots in many different schools across London, the oldest of which being Charing Cross Hospital Medical School which can be traced back to 1823 followed by teaching starting at Westminster Hospital in 1834 and St Mary’s Hospital in 1851.
In 1851 the Great Exhibition was organized as an exhibition of culture and industry by Henry Cole and by Prince Albert- husband of the reigning monarch of the United Kingdom Queen Victoria. An enormously popular and financial success proceeds from the Great Exhibition were designated to develop an area for cultural and scientific advancement in South Kensington. Within the next 6 years the Victoria and Albert Museum and Science Museum had opened joined by new facilities in 1871 for the Royal College of Chemistry and in 1881 for the Royal School of Mines; the opening of the Natural History Museum in 1881; and in 1888 the Imperial Institute.
In 1881 the Normal School of Science was established in South Kensington under the leadership of Thomas Huxley taking over responsibility for the teaching of the natural sciences and agriculture from the Royal School of Mines. The school was renamed the Royal College of Science by royal consent in 1890. The Central Institution of the City and Guilds of London Institute was opened as a technical education school on Exhibition Road by the Prince of Wales in early 1885.
20th century
At the start of the 20th century, there was a concern that Britain was falling behind Germany in scientific and technical education. A departmental committee was set up at the Board of Education in 1904, to look into the future of the Royal College of Science. A report released in 1906 called for the establishment of an institution unifying the Royal College of Science and the Royal School of Mines, as well as – if an agreement could be reached with the City and Guilds of London Institute – their Central Technical College.
On 8 July 1907 King Edward VII granted a Royal Charter establishing the Imperial College of Science and Technology. This incorporated the Royal School of Mines and the Royal College of Science. It also made provisions for the City and Guilds College to join once conditions regarding its governance were met as well as for Imperial to become a college of The University of London. The college joined the University of London on 22 July 1908 with the City and Guilds College joining in 1910. The main campus of Imperial College was constructed beside the buildings of the Imperial Institute- the new building for the Royal College of Science having opened across from it in 1906 and the foundation stone for the Royal School of Mines building being laid by King Edward VII in July 1909.
As students at Imperial had to study separately for London degrees in January 1919 students and alumni voted for a petition to make Imperial a university with its own degree awarding powers independent of the University of London. In response the University of London changed its regulations in 1925 so that the courses taught only at Imperial would be examined by the university enabling students to gain a BSc.
In October 1945 King George VI and Queen Elizabeth visited Imperial to commemorate the centenary of the Royal College of Chemistry which was the oldest of the institutions that united to form Imperial College. “Commemoration Day” named after this visit is held every October as the university’s main graduation ceremony. The college also acquired a biology field station at Silwood Park near Ascot, Berkshire in 1947.
Following the Second World War, there was again concern that Britain was falling behind in science – this time to the United States. The Percy Report of 1945 and Barlow Committee in 1946 called for a “British MIT”-equivalent backed by influential scientists as politicians of the time including Lord Cherwell; Sir Lawrence Bragg; and Sir Edward Appleton. The University Grants Committee strongly opposed however. So, a compromise was reached in 1953 where Imperial would remain within the university but double in size over the next ten years. The expansion led to a number of new buildings being erected. These included the Hill building in 1957 and the Physics building in 1960 and the completion of the East Quadrangle built in four stages between 1959 and 1965. The building work also meant the demolition of the City and Guilds College building in 1962–63 and the Imperial Institute’s building by 1967. Opposition from the Royal Fine Arts Commission and others meant that Queen’s Tower was retained with work carried out between 1966 and 1968 to make it free standing. New laboratories for biochemistry established with the support of a £350,000 grant from the Wolfson Foundation were opened by the Queen in 1965.
In 1988 Imperial merged with St Mary’s Hospital Medical School under the Imperial College Act 1988. Amendments to the royal charter changed the formal name of the institution to The Imperial College of Science Technology and Medicine and made St Mary’s a constituent college. This was followed by mergers with the National Heart and Lung Institute in 1995 and the Charing Cross and Westminster Medical School; Royal Postgraduate Medical School; and the Institute of Obstetrics and Gynecology in 1997 with the Imperial College Act 1997 formally establishing the Imperial College School of Medicine.
21st century
In 2003, Imperial was granted degree-awarding powers in its own right by the Privy Council. In 2004, the Imperial College Business School and a new main entrance on Exhibition Road were opened by Queen Elizabeth II. The UK Energy Research Centre was also established in 2004 and opened its headquarters at Imperial. On 9 December 2005, Imperial announced that it would commence negotiations to secede from the University of London. Imperial became fully independent of the University of London in July 2007.
In April 2011 Imperial and King’s College London joined the UK Centre for Medical Research and Innovation as partners with a commitment of £40 million each to the project. The centre was later renamed The Francis Crick Institute (UK) and opened on 9 November 2016. It is the largest single biomedical laboratory in Europe. The college began moving into the new White City campus in 2016 with the launching of the Innovation Hub. This was followed by the opening of the Molecular Sciences Research Hub for the Department of Chemistry officially opened by Mayor of London- Sadiq Khan in 2019. The White City campus also includes another biomedical centre funded by a £40 million donation by alumnus Sir Michael Uren.
Research
Imperial submitted a total of 1,257 staff across 14 units of assessment to the 2014 Research Excellence Framework (REF) assessment. This found that 91% of Imperial’s research is “world-leading” (46% achieved the highest possible 4* score) or “internationally excellent” (44% achieved 3*) giving an overall GPA of 3.36. In rankings produced by Times Higher Education based upon the REF results Imperial was ranked 2nd overall. Imperial is also widely known to have been a critical contributor of the discovery of penicillin; the invention of fiber optics; and the development of holography. The college promotes research commercialization partly through its dedicated technology transfer company- Imperial Innovations- which has given rise to a large number of spin-out companies based on academic research. Imperial College has a long-term partnership with the Massachusetts Institute of Technology that dates back from World War II. The United States is the college’s top collaborating foreign country with more than 15,000 articles co-authored by Imperial and U.S.-based authors over the last 10 years.
In January 2018 the mathematics department of Imperial and the CNRS-The National Center for Scientific Research[Centre national de la recherche scientifique](FR) launched UMI Abraham de Moivre at Imperial- a joint research laboratory of mathematics focused on unsolved problems and bridging British and French scientific communities. The Fields medallists Cédric Villani and Martin Hairer hosted the launch presentation. The CNRS-Imperial partnership started a joint PhD program in mathematics and further expanded in June 2020 to include other departments. In October 2018, Imperial College launched the Imperial Cancer Research UK Center- a research collaboration that aims to find innovative ways to improve the precision of cancer treatments inaugurated by former Vice President of the United States Joe Biden as part of his Biden Cancer Initiative.
Imperial was one of the ten leading contributors to the National Aeronautics and Space Administration InSight Mars lander which landed on planet Mars in November 2018, with the college logo appearing on the craft. InSight’s Seismic Experiment for Interior Structure, developed at Imperial, measured the first likely marsquake reading in April 2019. In 2019 it was revealed that the Blackett Laboratory would be constructing an instrument for the European Space (EU) Solar Orbiter in a mission to study the Sun, which launched in February 2020. The laboratory is also designing part of the DUNE/LBNF Deep Underground Neutrino Experiment.
In early 2020 immunology research at the Faculty of Medicine focused on SARS-CoV-2 under the leadership of Professor Robin Shattock as part of the college’s COVID-19 Response Team including the search of a cheap vaccine which started human trials on 15 June 2020. Professor Neil Ferguson’s 16 March report entitled Impact of non-pharmaceutical interventions (NPIs) to reduce COVID- 19 mortality and healthcare demand was described in a 17 March The New York Times article as the coronavirus “report that jarred the U.S. and the U.K. to action”. Since 18 May 2020 Imperial College’s Dr. Samir Bhatt has been advising the state of New York for its reopening plan. Governor of New York Andrew Cuomo said that “the Imperial College model- as we’ve been following this for weeks- was the best most accurate model.” The hospitals from the Imperial College Healthcare NHS Trust which have been caring for COVID-19 infected patients partnered with Microsoft to use their HoloLens when treating those patients reducing the amount of time spent by staff in high-risk areas by up to 83% as well as saving up to 700 items of PPE per ward, per week.
5.8.24
Jeremy P Rumsey
rumseyjp@ornl.gov
865.576.2038
Robots revolutionized the manufacturing industry by automating assembly lines. Now, more and more, they are being used to expedite the pace of scientific discovery.
Neutron scattering instruments are like giant high-powered microscopes that use beams of neutrons to study materials at the atomic scale. The BIO-SANS instrument, located at Oak Ridge National Laboratory’s High Flux Isotope Reactor, or HFIR, is the latest neutron scattering instrument to be retrofitted with state-of-the-art robotics and custom software.
The AI-driven HyperCT platform has three primary points of articulation that can rotate a sample in almost any direction, eliminating the need for human intervention and significantly reducing lengthy experiment times. Credit: Genevieve Martin, ORNL/U.S. Dept. of Energy
The sophisticated upgrade quadruples the number of samples the instrument can measure automatically and significantly reduces the need for human assistance. BIO-SANS specializes in studying the behavior, shape and size of complex biological materials.
“It’s a remarkable step forward in capabilities. The ability to analyze more samples means we’ll get better results,” said ORNL Group Leader Mark Loguillo. “Not only that, but automation provides us with better time management which, in turn, allows us to make more measurements in an experiment than we could before.”
Retrofitting robotics increases efficiency of neutron experiments
With a new robotic upgrade, BIO-SANS now measures samples more efficiently. The robot changes samples automatically, reducing the need for human assistance.
ORNL researcher Felicia Gilliland loads experiment samples into position for the newly installed UR5E robotic arm at the BIO-SANS instrument. The industrial-grade robot changes samples automatically, reducing the need for human assistance and improving sample throughput. Credit: Jeremy Rumsey/ORNL, U.S. Dept. of Energy
Installed on BIO-SANS is the UR5E Universal Robot, a lightweight mechanical arm equipped with five highly articulating joints. The robot is affixed to the ceiling inside an aluminum frame that contains a platform for storing experimental samples. Grippers attached to the end of the arm act like fingers that grab samples — powders or liquid solutions stored inside small metal canisters a couple cubic inches in size.
The robot moves samples to and from a tray directly beneath it and positions them into a beam of neutrons for analysis. About 84 samples can be stored inside the sample enclosure at a time. The robot can operate nonstop as long as the sample tray is full, which is one of the only parts of the experiment that requires human assistance once the experiment begins. Researchers can collect their data and monitor their experiment from their office or home.
“When the COVID-19 pandemic hit, we knew we needed a way to automate experiments so that they could be done remotely,” said Loguillo. “We installed a prototype robot that clearly demonstrated the value of integrating robotics and the potential it had, but eventually the prototype suffered from reliability issues. The new industrial-grade robot is highly customizable, which allows us to continue making improvements to its software and capabilities.”
Funding for the project was provided by the Department of Energy’s Biopreparedness Research Virtual Environment, or BRaVE, initiative.
Plans are in development to outfit the robot with artificial intelligence that will help reduce the time it takes for each experiment by eliminating unnecessary measurements. In turn, future upgrades will enable BIO-SANS to accommodate more complex experiments and increase scientific productivity.
In addition to Loguillo, team members involved with the project include ORNL researchers Mariano Ruiz-Rodriguez and John Wenzel. Based on the robot’s success, the team plans to upgrade other instruments, such as GP-SANS at HFIR and the MANDI diffractometer at the Spallation Neutron Source, or SNS.
HFIR and SNS are DOE Office of Science user facilities.
Established in 1942, The DOE’s Oak Ridge National Laboratory is the largest science and energy national laboratory in the Department of Energy system (by size) and third largest by annual budget. It is located in the Roane County section of Oak Ridge, Tennessee. Its scientific programs focus on materials, neutron science, energy, high-performance computing, systems biology and national security, sometimes in partnership with the state of Tennessee, universities and other industries.
ORNL has several of the world’s top supercomputers, including Summit, ranked by the TOP500 as Earth’s seventh-most powerful.
It hosts the Center for Nanophase Materials Sciences, the BioEnergy Science Center, and the Consortium for Advanced Simulation of Light Water Nuclear Reactors.
ORNL is managed by UT-Battelle for the Department of Energy’s Office of Science. DOE’s Office of Science is the single largest supporter of basic research in the physical sciences in the United States, and is working to address some of the most pressing challenges of our time.
Areas of research
ORNL conducts research and development activities that span a wide range of scientific disciplines. Many research areas have a significant overlap with each other; researchers often work in two or more of the fields listed here. The laboratory’s major research areas are described briefly below.
Chemical sciences – ORNL conducts both fundamental and applied research in a number of areas, including catalysis, surface science and interfacial chemistry; molecular transformations and fuel chemistry; heavy element chemistry and radioactive materials characterization; aqueous solution chemistry and geochemistry; mass spectrometry and laser spectroscopy; separations chemistry; materials chemistry including synthesis and characterization of polymers and other soft materials; chemical biosciences; and neutron science. Electron microscopy – ORNL’s electron microscopy program investigates key issues in condensed matter, materials, chemical and nanosciences. Nuclear medicine – The laboratory’s nuclear medicine research is focused on the development of improved reactor production and processing methods to provide medical radioisotopes, the development of new radionuclide generator systems, the design and evaluation of new radiopharmaceuticals for applications in nuclear medicine and oncology. Physics – Physics research at ORNL is focused primarily on studies of the fundamental properties of matter at the atomic, nuclear, and subnuclear levels and the development of experimental devices in support of these studies. Population – ORNL provides federal, state and international organizations with a gridded population database, called Landscan, for estimating ambient population. LandScan is a raster image, or grid, of population counts, which provides human population estimates every 30 x 30 arc seconds, which translates roughly to population estimates for 1 kilometer square windows or grid cells at the equator, with cell width decreasing at higher latitudes. Though many population datasets exist, LandScan is the best spatial population dataset, which also covers the globe. Updated annually (although data releases are generally one year behind the current year) offers continuous, updated values of population, based on the most recent information. Landscan data are accessible through GIS applications and a USAID public domain application called Population Explorer.
From left, J.D. Rice, Trevor Michelson and Chris Seck look at a monitor in Seck’s lab. The three are wearing safety glasses to protect against the laser beams used by the scanning vibrometer, which is helping Seck quantify vibration of an appliance in his lab. Carlos Jones/ORNL, U.S. Dept. of Energy
The last time this team used Oak Ridge National Laboratory’s 3D scanning vibrometer, it was to measure a gigantic composite panel.
This time, they’re measuring the vibration amplitudes of a cryogenic ion trap – around 10 nanometers, peak to peak.
“This is the first real test of the absolute limits of the machine,” said researcher Blake Van Hoy of the Isotope Science and Engineering Directorate’s Enrichment Science and Engineering Division, or ESED.
Van Hoy is the resident expert on the 3D scanning vibrometer. After he wrote a plant capital equipment proposal pitching it as a long-term infrastructure investment, the lab in 2021 purchased the specialized equipment, which uses visible or infrared lasers from three heads to build a geometric matrix, scattering light and assessing vibration to produce high-fidelity measurements of just about any object – enormous to tiny, any shape.
Its primary use at ORNL so far has been to help ESED with research and development. The composite panel, for example, was measured along with other types of panels to see which was best at reducing vibration energy.
But Van Hoy has advocated for more widespread use of the scanning vibrometer, which belongs to the lab, not specifically to ESED.
That’s why he was excited to hear from Chris Seck, a research scientist in the Computing and Computational Sciences Directorate’s Quantum Sensing and Computing group.
Trevor Michelson and Chris Seck sit at a setup in Seck’s lab, where the cryogenic ion trap apparatus, center, is surrounded by three heads of the laser scanning vibrometer, each fixed on a different point. The vibrometer will help Seck quantify vibration on the apparatus. Carlos Jones/ORNL, U.S. Dept. of Energy
Seck is leading a project to engineer and develop a cryogenic ion trap apparatus to simulate quantum spin liquids, a key research area in materials science and neutron scattering studies. In the simulator, Seck can manipulate the trapped ion qubits – or quantum bits, the basic unit of information in quantum computing − to behave similarly to quantum materials that would be difficult to study in a lab.
But a common source of error in using cryogenic ion traps is the vibration of the actual apparatus, which factors into how the ions behave.
Seck is using a custom-built vacuum system inside a commercial cryo cooler to cool the apparatus, but he needs to be able to quantify the vibration of the mechanical cooler. When Seck read about the lab’s laser scanning vibrometer, he thought it could be used to quantify the vibrations. Not only would that give him a baseline for his own research, it also could help with engineering of future systems to further reduce vibrations.
“But this is much smaller than what they’re used to measuring,” Seck said.
It’s also different in other ways. For one, the team – which also includes ESED’s Trevor Michelson and J.D. Rice − will shoot at the tiny target area through glass. And while often the three laser heads are pointed at a single target, to provide a 3D model, in this case each head will point to an individual measurement point.
“This is going to be an interesting experiment for us because it’s pushing the absolute limits of everything we could think of to do,” Van Hoy said. “It’s going to be a good learning experience to know what the limits are.”
Many of the exceedingly detailed measurements the system can perform were once attempted with accelerometers, expensive and fragile button-size sensors that were placed in multiple locations to measure vibration frequency and damping.
Three scanning heads each are pointed toward a different measurement point. Carlos Jones/ORNL, U.S. Dept. of Energy
But in this case, that would never be an option, Michelson said, because the accelerometers would be too big to use in the small space, and their added weight would invalidate the experiment by changing the dynamic behavior of the ion trap and associated vacuum chamber.
If not for the availability of the vibrometer, which recently had been calibrated, Seck would have had to order parts and construct an optical beamline to measure the vibrations, investing both time and money. But Michelson and Rice were easily able to set up the vibrometer system in his Building 5700 lab.
Since Seck’s is a long-term project, Michelson and Rice said the vibrometer can be moved if it’s needed for another project in the meantime; it can be set up again in Seck’s lab in a matter of hours. Seck said he hopes the measurements, besides informing his research, will lead to an article in the Review of Scientific Instruments.
And the team said they hope increased work for the vibrometer will lead to a dedicated lab space that specifically can accommodate measuring any size – enormous to tiny.
“What’s fascinating to me is the versatility of this equipment,” Rice said. “The first thing we ever did out of the gate with it was a large plate from 12 feet away – but I can take the mechanical pencil and ping it and get a measurement almost to the tip. Literally everything from tiny little components, all the way up to big structures, as long as you can get a good line of sight between the laser and what you need to test, you can get incredibly detailed measurements.
“We’re still barely scratching the surface of its capabilities, and it’s already blowing me away.”
Established in 1942, The DOE’s Oak Ridge National Laboratory is the largest science and energy national laboratory in the Department of Energy system (by size) and third largest by annual budget. It is located in the Roane County section of Oak Ridge, Tennessee. Its scientific programs focus on materials, neutron science, energy, high-performance computing, systems biology and national security, sometimes in partnership with the state of Tennessee, universities and other industries.
ORNL has several of the world’s top supercomputers, including Summit, ranked by the TOP500 as Earth’s seventh-most powerful.
It hosts the Center for Nanophase Materials Sciences, the BioEnergy Science Center, and the Consortium for Advanced Simulation of Light Water Nuclear Reactors.
ORNL is managed by UT-Battelle for the Department of Energy’s Office of Science. DOE’s Office of Science is the single largest supporter of basic research in the physical sciences in the United States, and is working to address some of the most pressing challenges of our time.
Areas of research
ORNL conducts research and development activities that span a wide range of scientific disciplines. Many research areas have a significant overlap with each other; researchers often work in two or more of the fields listed here. The laboratory’s major research areas are described briefly below.
Chemical sciences – ORNL conducts both fundamental and applied research in a number of areas, including catalysis, surface science and interfacial chemistry; molecular transformations and fuel chemistry; heavy element chemistry and radioactive materials characterization; aqueous solution chemistry and geochemistry; mass spectrometry and laser spectroscopy; separations chemistry; materials chemistry including synthesis and characterization of polymers and other soft materials; chemical biosciences; and neutron science. Electron microscopy – ORNL’s electron microscopy program investigates key issues in condensed matter, materials, chemical and nanosciences. Nuclear medicine – The laboratory’s nuclear medicine research is focused on the development of improved reactor production and processing methods to provide medical radioisotopes, the development of new radionuclide generator systems, the design and evaluation of new radiopharmaceuticals for applications in nuclear medicine and oncology. Physics – Physics research at ORNL is focused primarily on studies of the fundamental properties of matter at the atomic, nuclear, and subnuclear levels and the development of experimental devices in support of these studies. Population – ORNL provides federal, state and international organizations with a gridded population database, called Landscan, for estimating ambient population. LandScan is a raster image, or grid, of population counts, which provides human population estimates every 30 x 30 arc seconds, which translates roughly to population estimates for 1 kilometer square windows or grid cells at the equator, with cell width decreasing at higher latitudes. Though many population datasets exist, LandScan is the best spatial population dataset, which also covers the globe. Updated annually (although data releases are generally one year behind the current year) offers continuous, updated values of population, based on the most recent information. Landscan data are accessible through GIS applications and a USAID public domain application called Population Explorer.
Astronomers, using the Five-hundred-meter Aperture Spherical radio Telescope (FAST) in China’s Guizhou Province, have found an abundance of gas-rich galaxies in the distant universe.
Dr. XI Hongwei from the National Astronomical Observatories of the Chinese Academy of Sciences (NAOC), and the collaborators, revealed the properties of six new high-redshift galaxies. The study was published online in The Astrophysical Journal Letters on May 10.
These remarkable galaxies, whose radio wave emissions have taken almost the age of the solar system to reach us, contain similar or more atomic hydrogen gas than the tens of thousands of galaxies previously surveyed in the local universe with other radio telescopes.
The researchers concluded that galaxies four billion years ago have much more star-forming gas than current day galaxies, and that distant galaxies have much greater gas reservoirs than previously believed.
“These discoveries are part of the ongoing FAST Ultra Deep Survey, showing the tremendous sensitivity of the world’s largest radio telescope,” said Prof. PENG Bo from NAOC, one of the corresponding authors. “The new FAST survey has so far discovered over 100 new galaxies at distances up to five billion light years, with the final number expected to reach over 1000.”
Finding the optical counterparts to the new radio discoveries has turned into a detective story, because galaxies are very faint at such large distances. And because of the wavelength difference, the localization accuracy of FAST is not as good as that of optical telescopes.
However, using the largest optical telescopes in US and Russia, the counterparts were eventually identified by experts from the collaborative team. The counterparts were found to contain 2-3 times more stars than the Milky Way, yet contain about 10 times the mass of hydrogen gas.
“This collaborative work between Chinese and Australia radio astronomers demonstrates the tremendous potential of the new generation of radio telescopes that, later this decade, will also include the international Square Kilometre Array Observatory (SKAO),” said Prof. Lister Staveley-Smith from the University of Western Australia node of the International Centre for Radio Astronomy research, the other corresponding author.
Figure 1: Signals of the new six galaxies discovered by FAST (black lines) and confirmed by optical telescopes (cyan lines). (Image by NAOC)
Figure 2: Radio contours (white lines) overlayed on optical images. Their optical counterparts are zoomed in at bottom right corners. Red circles show the FAST resolution. (Image by NAOC)
The Chinese Academy of Sciences[中国科学院](CN) is the national academy for the natural sciences of the People’s Republic of China. It has historical origins in the Academia Sinica during the Republican era and was formerly also known by that name. Collectively known as the “Two Academies (两院)” along with the Chinese Academy of Engineering, it functions as the national scientific think tank and academic governing body, providing advisory and appraisal services on issues stemming from the national economy, social development, and science and technology progress. It is headquartered in Xicheng District, Beijing, with branch institutes all over mainland China. It has also created hundreds of commercial enterprises, Lenovo being one of the most famous.
The Chinese Academy of Sciences has been ranked the No. 1 research institute in the world by Nature Index since the list’s inception in 2016 by Nature Portfolio. It is the most productive institution publishing articles of sustainable development indexed in Web of Science among all universities and research institutions in the world.
The Chinese Academy of Sciences has six academic divisions:
Chemistry (化学部)
Information Technological Sciences (信息技术科学部)
Earth Sciences (地学部)
Life Sciences and Medical Sciences (生命科学和医学学部)
Mathematics and Physics (数学物理学部)
Technological Sciences (技术科学部)
The CAS has thirteen regional branches, in Beijing, Shenyang, Changchun, Shanghai, Nanjing, Wuhan, Guangzhou, Chengdu, Kunming, Xi’an, Lanzhou, Hefei and Xinjiang. It has over one hundred institutes and four universities (the University of Science and Technology of China at Hefei, Anhui, the University of the Chinese Academy of Sciences in Beijing, ShanghaiTech University, and Shenzhen Institute of Advanced Technology). Backed by the institutes of CAS, UCAS is headquartered in Beijing, with graduate education bases in Shanghai, Chengdu, Wuhan, Guangzhou and Lanzhou, four Science Libraries of Chinese Academy of Sciences, three technology support centers and two news and publishing units. These CAS branches and offices are located in 20 provinces and municipalities throughout China. CAS has invested in or created over 430 science- and technology-based enterprises in eleven industries, including eight companies listed on stock exchanges.
Being granted a Fellowship of the Academy represents the highest level of national honor for Chinese scientists. The CAS membership system includes Academicians (院士), Emeritus Academicians (荣誉院士) and Foreign Academicians (外籍院士).
The Chinese Academy of Sciences was ranked very highly in the Nature Index Annual Tables, which measure the largest contributors to papers published in 82 leading journals.
Research institutes
Beijing Branch
University of the Chinese Academy of Sciences (UCAS)
Academy of Mathematics and Systems Science
Institute of Acoustics (IOA)
Institute of Atmospheric Physics
Institute of Botany, Chinese Academy of Sciences
Institute of Physics (IOPCAS)
Institute of Semiconductors
Institute of Electrical Engineering (IEE)
Institute of Information Engineering (IIE)
Institute of Theoretical Physics
Institute of High Energy Physics
Institute of Biophysics
Institute of Genetics and Developmental Biology
Institute of Electronics
National Astronomical Observatories
Institute of Computing Technology
Institute of Software
Institute of Automation
Beijing Institute of Genomics
Institute of Geographic Sciences and Natural Resources
Institute of Geology and Geophysics (IGG)
Institute of Remote Sensing and Digital Earth
Institute of Tibetan Plateau Research
Institute of Vertebrate Paleontology and Paleoanthropology
National Center for Nanoscience and Technology
Institute of Policy and Management
Institute of Psychology
Institute of Zoology
Changchun Branch
Changchun Institute of Optics, Fine Mechanics and Physics
Changchun Institute of Applied Chemistry
Northeast Institute of Geography and Agroecology
Changchun Observatory
Chengdu Branch
Institute of Mountain Hazards and Environment
Chengdu Institute of Biology
Institute of Optics and Electronics
Chengdu Institute of Organic Chemistry
Institute of Computer Application
Chongqing Institute of Green and Intelligent Technology
Guangzhou Branch
South China Botanical Garden
Shenzhen Institutes of Advanced Technology
South China Sea Institute of Oceanology
Guangzhou Institute of Energy Conversion
Guangzhou Institute of Geochemistry
Guangzhou Institute of Biomedicine and Health
Guiyang Branch
Institute of Geochemistry
Hefei Branch
Hefei Institutes of Physical Science
University of Science and Technology of China
Kunming Branch
Kunming Institute of Botany
Kunming Institute of Zoology
Xishuangbanna Tropical Botanical Garden
Institute of Geochemistry
Yunnan Astronomical Observatory
Lanzhou Branch
Institute of Modern Physics
Lanzhou Institute of Chemical Physics
Lanzhou Institute of Geology
Northwest Institute of Plateau Biology
Northwest Institute of Eco-Environment and Resources
Qinghai Institute of Salt Lakes Research
Nanjing Branch
Purple Mountain Observatory (Zijinshan Astronomical Observatory)
Institute of Soil Science
Nanjing Institute of Geology and Palaeontology
Nanjing Institute of Geography and Limnology
Nanjing Institute of Astronomical Optics and Technology
Suzhou Institute of Nano-tech and Nano-bionics (SINANO)
Suzhou Institute of Biomedical Engineering and Technology (SIBET)
Nanjing Botanical Garden, Memorial Sun Yat-Sen (Institute of Botany, Jiangsu Province and Chinese Academy of Science)
University of Chinese Academy of Sciences, Nanjing College
Shanghai Branch
Shanghai Astronomical Observatory
Shanghai Institute of Microsystem and Information Technology
Shanghai Institute of Technical Physics
Shanghai Institute of Optics and Fine Mechanics
Shanghai Institute of Ceramics
Shanghai Institute of Organic Chemistry
Shanghai Institute of Applied Physics
Shanghai Institutes for Biological Sciences
Shanghai Institute of Materia Medica
Institut Pasteur of Shanghai
Shanghai Advanced Research Institute, CAS
Institute of Neuroscience (ION)
ShanghaiTech University
Shenyang Branch
Institute of Metal Research
Shenyang Institute of Automation
Shenyang Institute of Applied Ecology, formerly the Institute of Forestry and Pedology
Shenyang Institute of Computing Technology
Dalian Institute of Chemical Physics
Qingdao Institute of Oceanology
Qingdao Institute of Bioenergy and Bioprocess Technology
Yantai Institute of Coastal Zone Research
Taiyuan Branch
Shanxi Institute of Coal Chemistry (ICCCAS)
Wuhan Branch
Wuhan Institute of Rock and Soil Mechanics
Wuhan Institute of Physics and Mathematics
Wuhan Institute of Virology
Institute of Geodesy and Geophysics
Institute of Hydrobiology
Wuhan Botanical Garden
Xinjiang Branch
Xinjiang Technical Institute of Physics and Chemistry
Xinjiang Institute of Ecology and Geography
Xi’an Branch
Xi’an Institute of Optics and Precision Mechanics
National Time Service Center
Institute of Earth Environment
A collage of artist concepts highlighting the novel approaches proposed by the 2024 NIAC Phase II awardees for possible future missions. Credits: NASA, From left: Edward Balaban, Mary Knapp, Mahmooda Sultana, Brianna Clements, Ethan Schaler
NASA’s Innovative Advanced Concepts program (NIAC) has selected six visionary concept studies for additional funding and development. Each study has already completed the initial NIAC phase, showing their futuristic ideas – like a lunar railway system and fluid-based telescopes – may provide fresh perspectives and approaches as NASA explores the unknown in space.
The NIAC Phase II conceptual studies will receive up to $600,000 to continue working over the next two years to address key remaining technical and budget hurdles and pave their development path forward. When Phase II is complete, these studies could advance to the final NIAC phase, earning additional funding and development consideration toward becoming a future aerospace mission.
“These diverse, science fiction-like concepts represent a fantastic class of Phase II studies,” said John Nelson, NIAC program executive at NASA Headquarters in Washington. “Our NIAC fellows never cease to amaze and inspire, and this class definitely gives NASA a lot to think about in terms of what’s possible in the future.” The six concepts chosen for 2024 NIAC Phase II awards are:
Fluidic Telescope (FLUTE): Enabling the Next Generation of Large Space Observatories would create a large optical observatory in space using fluidic shaping of ionic liquids. These in-space observatories could potentially help investigate NASA’s highest priority astrophysics targets, including Earth-like exoplanets, first-generation stars, and young galaxies. The FLUTE study is led by Edward Balaban from NASA’s Ames Research Center in California’s Silicon Valley.
Pulsed Plasma Rocket: Shielded, Fast Transits for Humans to Mars is an innovative propulsion system that relies on using fission-generated packets of plasma for thrust. This innovative system could significantly reduce travel times between Earth and any destination in the solar system. This study is led by Brianna Clements with Howe Industries in Scottsdale, Arizona.
The Great Observatory for Long Wavelengths (GO-LoW) could change the way NASA conducts astronomy. This mega constellation low-frequency radio telescope uses thousands of autonomous SmallSats capable of measuring the magnetic fields emitted from exoplanets and the cosmic dark ages. GO-LoW is led by Mary Knapp with MIT in Cambridge, Massachusetts.
Radioisotope Thermoradiative Cell Power Generator is investigating new in-space power sources, potentially operating at higher efficiencies than NASA legacy power generators. This technology could enable small exploration and science spacecraft in the future that are unable to carry bulky solar or nuclear power systems. This power generation concept study is from Stephen Polly at the Rochester Institute of Technology in New York.
FLOAT: Flexible Levitation on a Track would be a lunar railway system, providing reliable, autonomous, and efficient payload transport on the Moon. This rail system could support daily operations of a sustainable lunar base as soon as the 2030s. Ethan Schaler leads FLOAT at NASA’s Jet Propulsion Laboratory in Southern California.
ScienceCraft for Outer Planet Exploration distributes Quantum Dot-based sensors throughout the surface of a solar sail, enabling it to become an innovative imager. Quantum physics would allow NASA to take scientific measurements through studying how the dots absorb light. By leveraging the solar sail’s area, it allows lighter, more cost-effective spacecraft to carry imagers across the solar system. ScienceCraft is led by NASA’s Mahmooda Sultana at the agency’s Goddard Space Flight Center in Greenbelt, Maryland.
NASA’s Space Technology Mission Directorate funds the NIAC program, as it is responsible for developing the agency’s new cross-cutting technologies and capabilities to achieve its current and future missions.
To learn more about NIAC and the 2024 Phase II studies, visit:
The National Aeronautics and Space Administration is the agency of the United States government that is responsible for the nation’s civilian space program and for aeronautics and aerospace research.
President Dwight D. Eisenhower established the National Aeronautics and Space Administration (NASA) in 1958 with a distinctly civilian (rather than military) orientation encouraging peaceful applications in space science. The National Aeronautics and Space Act was passed on July 29, 1958, disestablishing NASA’s predecessor, the National Advisory Committee for Aeronautics (NACA). The new agency became operational on October 1, 1958.
Since that time, most U.S. space exploration efforts have been led by NASA, including the Apollo moon-landing missions, the Skylab space station, and later the Space Shuttle. Currently, NASA is supporting the International Space Station and is overseeing the development of the Orion Multi-Purpose Crew Vehicle and Commercial Crew vehicles. The agency is also responsible for the Launch Services Program (LSP) which provides oversight of launch operations and countdown management for unmanned NASA launches. Most recently, NASA announced a new Space Launch System that it said would take the agency’s astronauts farther into space than ever before and lay the cornerstone for future human space exploration efforts by the U.S.
And now NASA’s PACE spacecraft will help us better understand our ocean and atmosphere by measuring key variables associated with cloud formation, particles and pollutants in the air, and microscopic, floating marine life (phytoplankton). These observations will help us better monitor ocean health, air quality, and climate change.
5.10.24
Gerelle Dodson
Headquarters, Washington
202-358-4637
gerelle.q.dodson@nasa.gov
A photo of a team of researchers from the University of Puerto Rico-Río Piedras while working to discover a more efficient water recycling system for use on space missions. The team is comprised of doctoral students Liz Santiago-Martoral, on the left, and Alondra Rodriguez-Rolon, and their mentor Professor Eduardo Nicolau. One of their experiments can be seen on the countertop to the left of the group. Credit: NASA
NASA is awarding approximately $45 million to 21 higher-education institutions to help build capacity for research. The awards were made possible through the Minority University Research and Education Project Institutional Research Opportunity (MIRO) and Established Program to Stimulate Competitive Research (EPSCoR) grants, which are funded by the agency’s Office of Science, Technology, Engineering, and Mathematics (STEM) Engagement.
“NASA’s Minority University Research and Education Project Institutional Research Opportunity and Established Program to Stimulate Competitive Research awards help institutions raise their technological bar,” said Torry Johnson, deputy associate administrator of STEM Engagement Programs at NASA Headquarters in Washington. “When institutions enhance their capabilities and infrastructure, they become more competitive in their research, which opens doors to valuable experience and opportunities.”
Minority University Research and Education Project Institutional Research Opportunity (MIRO) Awards
Seven minority-serving institutions will receive approximately $5 million each over a five-year period of performance for projects that span a variety of research topics. The institutions and their proposed projects are:
Alaska Pacific University in Anchorage – Alaska Pacific University Microplastics Research and Education Center California State University in Fullerton – SpaceIgnite Center for Advanced Research-Education in Combustion City University of New York, Hunter College in New York – NASA-Hunter College Center for Advanced Energy Storage for Space Florida Agricultural and Mechanical University in Tallahassee – Integrative Space Additive Manufacturing: Opportunities for Workforce-Development in NASA Related Materials Research and Education New Jersey Institute of Technology in Newark – AI Powered Solar Eruption Center of Excellence in Research and Education University of Houston in Houston – NASA MIRO Inflatable Deployable Environment and Adaptive Space Systems Center University of Illinois in Chicago – Center for In-Space Manufacturing: Recycling and Regolith Processing
NASA’s MIRO award was established to strengthen and develop research capacity and infrastructure of minority serving institutions in areas of strategic importance and value to NASA missions and national priorities.
Established Program to Stimulate Competitive Research (EPSCoR) Award
NASA establishes partnerships with government, higher education, and industry to create lasting improvements in research infrastructure and capacity for specific states or regions, while enhancing its national research and development competitiveness. The program is directed at those jurisdictions that have traditionally not participated in competitive aerospace and aerospace-related research activities.
NASA will award 14 institutions up to $750,000 each over the course of a three-year period of performance. The awarded institutions and their projects are:
University of Mississippi in University – Development of a Lagrangian Stability Analysis Framework for High-Speed Boundary Layers University of Alabama in Huntsville – Testing the functionality and performance of a large area detector for STROBE-X Louisiana State University in Baton Rouge – Colloidal Assembly: Understanding the Electric Field Driven Assembly of Colloids and its Applications (Science Mission Directorate) West Virginia University in Morgantown – Science Mission Directorate: Bringing Gravitational-Wave Astronomy into the Space Age: Next-Generation Waveform Modeling of Black-Hole Binary Coalescences for Laser Intererometer Space Antenna Data Analysis University of Puerto Rico in San Juan – NASA EPSCoR: Space Technology Mission Directorate/Jet Propulsion Laboratory: Advancing High-Energy, Cycle-Stable Sulfur-Based Batteries for NASA Space Missions: An Integrated Framework of Density Functional Theory, Machine Learning, and Materials Innovation Desert Research Institute, Reno, Nevada – NASA’s Ames Research Center in Silicon Valley, California: Prospecting and Pre-Colonization of the Moon and Mars using Autonomous Robots with Human-In-The-Loop Oklahoma State University in Stillwater – A.7.4.2 Biosignature Detection of Solar System Ocean Worlds using Science-Guided Machine Learning Iowa State University in Ames – Johnson Space Center, Ames Research Center: Non-GPS Navigation System Using Dual Star/Planetary Cameras for Lunar and Deep-Space CubeSat Missions University of Alaska Fairbanks in Fairbanks – NASA’s Glenn Research Center in Cleveland: The Alaska – Venus analog: synthesizing seismic ground motion and wind noise in extreme environments University of the Virgin Islands in Charlotte Amalie – University of the Virgin Islands Etelman Observatory in the Era of Time Domain and MultiMessenger Astronomy: Preparing for a New Era of Science Productivity University of Hawaii at Manoa in Honolulu – Cubesats for Climate Change Detection of Transient Greenhouse Gas Emissions University of Idaho in Moscow – Science Mission Directorate and Goddard Space Flight Center: Improving Global Dryland Streamflow Modeling by Better Characterizing Vegetation Use of Deep-Water Resources Using NASA’s Gravity Recovery and Climate Experiment/Gravity Recovery and Climate Experiment Follow-On, SWOT, and Land Information System University of Arkansas in Little Rock – AR- III-Nitride Ultraviolet Laser Diodes for Harsh Environments, Space Based Communications, and Remote Sensing (Space Technology Mission Directorate) South Dakota School of Mines and Technology in Rapid City – Science Mission Directorate: High Spatial-Temporal Resolution Soil Moisture Retrieval using Deep Learning Fusion of Multimodal Satellite Datastreams
Both awards were made through NASA’s Office of STEM engagement solicitations. They promote STEM literacy to enhance and sustain the capability of institutions to perform NASA-related research and education, which directly supports the agency’s mission directorates.
The National Aeronautics and Space Administration is the agency of the United States government that is responsible for the nation’s civilian space program and for aeronautics and aerospace research.
President Dwight D. Eisenhower established the National Aeronautics and Space Administration (NASA) in 1958 with a distinctly civilian (rather than military) orientation encouraging peaceful applications in space science. The National Aeronautics and Space Act was passed on July 29, 1958, disestablishing NASA’s predecessor, the National Advisory Committee for Aeronautics (NACA). The new agency became operational on October 1, 1958.
Since that time, most U.S. space exploration efforts have been led by NASA, including the Apollo moon-landing missions, the Skylab space station, and later the Space Shuttle. Currently, NASA is supporting the International Space Station and is overseeing the development of the Orion Multi-Purpose Crew Vehicle and Commercial Crew vehicles. The agency is also responsible for the Launch Services Program (LSP) which provides oversight of launch operations and countdown management for unmanned NASA launches. Most recently, NASA announced a new Space Launch System that it said would take the agency’s astronauts farther into space than ever before and lay the cornerstone for future human space exploration efforts by the U.S.
And now NASA’s PACE spacecraft will help us better understand our ocean and atmosphere by measuring key variables associated with cloud formation, particles and pollutants in the air, and microscopic, floating marine life (phytoplankton). These observations will help us better monitor ocean health, air quality, and climate change.
Results after closing the Cycle 11 Call for Proposals (CfP) shows the community submitted 1,713 proposals to make observations with ALMA, marking a slight increase from the previous year’s Cycle 10 (1,679), thus maintaining consistency in request numbers (see fig. 1). Each proposal will now go through an anonymous selection process to determines which projects will be observed in this cycle.
The amount of requested observing time for the 12-m array continues to increase steadily: 31,751 hours stands as the most time ever requested in a single cycle (see fig. 2). The amount of observing time requested is 7.4 times larger than the amount of time available.
Even though the amount of time requested for the Morita Array (in both 7-m array and Total Power array) decreased, the number of submitted proposals for it is approximately the same as recent cycles, with a strong oversubscription of 3.0 on the 7-m array. This means there were fewer proposals requesting very large amounts of observation time for both the 7-m (with approximately 14,000 hours) and Total Power (with 9,000 hours) arrays this year, in comparison to the previous Cycle 10 (see fig. 2).
The community interest in Large Programs remains strong, as 42 proposals were submitted for 4,700 hours on the 12-m array, close to the previous year record, with 44 proposals requesting nearly 5,000 hours. The 7-m and TP arrays received 2,200 and 1,300 hours of observation requests respectively.
The community responded enthusiastically once again to the Joint Proposal opportunity. A total of 67 Joint Proposals were submitted, which is a 60% increase over Cycle 10 (with 42 proposals). In total, 50 Joint Proposals requested time on James Webb Space Telescope (almost twice than previous cycle), 14 requested NRAO’s Karl G. Jansky Very Large Array (VLA), and 5 requested the European Southern Observatory’s Very Large Telescope (VLT).
“The continued strong demand for ALMA time is a testament to the great science ALMA has carried out to date. We look forward to the exciting science and the new discoveries in Cycle 11.”, says John Carpenter, ALMA Observatory Scientist.
The proposals have been distributed to the reviewers, and the results of the review process will be distributed by early August.
The antennas can be moved across the desert plateau over distances from 150 m to 16 km, which will give ALMA a powerful variable “zoom”, similar in its concept to that employed at the centimetre-wavelength Very Large Array (VLA) site in New Mexico, United States.
The high sensitivity is mainly achieved through the large numbers of antenna dishes that will make up the array.
The telescopes were provided by the European, North American and East Asian partners of ALMA. The American and European partners each provided twenty-five 12-meter diameter antennas, that compose the main array. The participating East Asian countries are contributing 16 antennas (four 12-meter diameter and twelve 7-meter diameter antennas) in the form of the Atacama Compact Array (ACA), which is part of the enhanced ALMA.
By using smaller antennas than the main ALMA array, larger fields of view can be imaged at a given frequency using ACA. Placing the antennas closer together enables the imaging of sources of larger angular extent. The ACA works together with the main array in order to enhance the latter’s wide-field imaging capability.
ALMA has its conceptual roots in three astronomical projects — the Millimeter Array (MMA) of the United States, the Large Southern Array (LSA) of Europe, and the Large Millimeter Array (LMA) of Japan.
The first step toward the creation of what would become ALMA came in 1997, when the National Radio Astronomy Observatory (NRAO) and the European Southern Observatory (ESO) agreed to pursue a common project that merged the MMA and LSA. The merged array combined the sensitivity of the LSA with the frequency coverage and superior site of the MMA. ESO and NRAO worked together in technical, science, and management groups to define and organize a joint project between the two observatories with participation by Canada and Spain (the latter became a member of ESO later).
A series of resolutions and agreements led to the choice of “Atacama Large Millimeter Array”, or ALMA, as the name of the new array in March 1999 and the signing of the ALMA Agreement on 25 February 2003, between the North American and European parties. (“Alma” means “soul” in Spanish and “learned” or “knowledgeable” in Arabic.) Following mutual discussions over several years, the ALMA Project received a proposal from the National Astronomical Observatory of Japan (NAOJ) whereby Japan would provide the ACA (Atacama Compact Array) and three additional receiver bands for the large array, to form Enhanced ALMA. Further discussions between ALMA and NAOJ led to the signing of a high-level agreement on 14 September 2004 that makes Japan an official participant in Enhanced ALMA, to be known as the Atacama Large Millimeter/submillimeter Array. A groundbreaking ceremony was held on November 6, 2003 and the ALMA logo was unveiled.
During an early stage of the planning of ALMA, it was decided to employ ALMA antennas designed and constructed by known companies in North America, Europe, and Japan, rather than using one single design. This was mainly for political reasons. Although very different approaches have been chosen by the providers, each of the antenna designs appears to be able to meet ALMA’s stringent requirements. The components designed and manufactured across Europe were transported by specialist aerospace and astrospace logistics company Route To Space Alliance, 26 in total which were delivered to Antwerp for onward shipment to Chile.
A new machine-learning technique can train and control a reconfigurable soft robot that can dynamically change its shape to complete a task. The researchers, from MIT and elsewhere, also built a simulator that can evaluate control algorithms for shape-shifting soft robots. Image: Courtesy of the researchers; MIT News
Imagine a slime-like robot that can seamlessly change its shape to squeeze through narrow spaces, which could be deployed inside the human body to remove an unwanted item.
While such a robot does not yet exist outside a laboratory, researchers are working to develop reconfigurable soft robots for applications in health care, wearable devices, and industrial systems.
But how can one control a squishy robot that doesn’t have joints, limbs, or fingers that can be manipulated, and instead can drastically alter its entire shape at will? MIT researchers are working to answer that question.
They developed a control algorithm that can autonomously learn how to move, stretch, and shape a reconfigurable robot to complete a specific task, even when that task requires the robot to change its morphology multiple times. The team also built a simulator to test control algorithms for deformable soft robots on a series of challenging, shape-changing tasks.
Their method completed each of the eight tasks they evaluated while outperforming other algorithms. The technique worked especially well on multifaceted tasks. For instance, in one test, the robot had to reduce its height while growing two tiny legs to squeeze through a narrow pipe, and then un-grow those legs and extend its torso to open the pipe’s lid.
While reconfigurable soft robots are still in their infancy, such a technique could someday enable general-purpose robots that can adapt their shapes to accomplish diverse tasks.
“When people think about soft robots, they tend to think about robots that are elastic, but return to their original shape. Our robot is like slime and can actually change its morphology. It is very striking that our method worked so well because we are dealing with something very new,” says Boyuan Chen, an electrical engineering and computer science (EECS) graduate student and co-author of a paper on this approach.
Chen’s co-authors include lead author Suning Huang, an undergraduate student at Tsinghua University in China who completed this work while a visiting student at MIT; Huazhe Xu, an assistant professor at Tsinghua University; and senior author Vincent Sitzmann, an assistant professor of EECS at MIT who leads the Scene Representation Group in the Computer Science and Artificial Intelligence Laboratory. The research will be presented at the International Conference on Learning Representations.
Controlling dynamic motion
Scientists often teach robots to complete tasks using a machine-learning approach known as reinforcement learning, which is a trial-and-error process in which the robot is rewarded for actions that move it closer to a goal.
This can be effective when the robot’s moving parts are consistent and well-defined, like a gripper with three fingers. With a robotic gripper, a reinforcement learning algorithm might move one finger slightly, learning by trial and error whether that motion earns it a reward. Then it would move on to the next finger, and so on.
But shape-shifting robots, which are controlled by magnetic fields, can dynamically squish, bend, or elongate their entire bodies.
The researchers built a simulator to test control algorithms for deformable soft robots on a series of challenging, shape-changing tasks. Here, a reconfigurable robot learns to elongate and curve its soft body to weave around obstacles and reach a target. Image: Courtesy of the researchers
“Such a robot could have thousands of small pieces of muscle to control, so it is very hard to learn in a traditional way,” says Chen.
To solve this problem, he and his collaborators had to think about it differently. Rather than moving each tiny muscle individually, their reinforcement learning algorithm begins by learning to control groups of adjacent muscles that work together.
Then, after the algorithm has explored the space of possible actions by focusing on groups of muscles, it drills down into finer detail to optimize the policy, or action plan, it has learned. In this way, the control algorithm follows a coarse-to-fine methodology.
“Coarse-to-fine means that when you take a random action, that random action is likely to make a difference. The change in the outcome is likely very significant because you coarsely control several muscles at the same time,” Sitzmann says.
To enable this, the researchers treat a robot’s action space, or how it can move in a certain area, like an image.
Their machine-learning model uses images of the robot’s environment to generate a 2D action space, which includes the robot and the area around it. They simulate robot motion using what is known as the material-point-method, where the action space is covered by points, like image pixels, and overlayed with a grid.
The same way nearby pixels in an image are related (like the pixels that form a tree in a photo), they built their algorithm to understand that nearby action points have stronger correlations. Points around the robot’s “shoulder” will move similarly when it changes shape, while points on the robot’s “leg” will also move similarly, but in a different way than those on the “shoulder.”
In addition, the researchers use the same machine-learning model to look at the environment and predict the actions the robot should take, which makes it more efficient.
Building a simulator
After developing this approach, the researchers needed a way to test it, so they created a simulation environment called DittoGym.
DittoGym features eight tasks that evaluate a reconfigurable robot’s ability to dynamically change shape. In one, the robot must elongate and curve its body so it can weave around obstacles to reach a target point. In another, it must change its shape to mimic letters of the alphabet.
In this simulation, the reconfigurable soft robot, trained using the researchers’ control algorithm, must change its shape to mimic objects, like stars, and the letters M-I-T. Image: Courtesy of the researchers
“Our task selection in DittoGym follows both generic reinforcement learning benchmark design principles and the specific needs of reconfigurable robots. Each task is designed to represent certain properties that we deem important, such as the capability to navigate through long-horizon explorations, the ability to analyze the environment, and interact with external objects,” Huang says. “We believe they together can give users a comprehensive understanding of the flexibility of reconfigurable robots and the effectiveness of our reinforcement learning scheme.”
Their algorithm outperformed baseline methods and was the only technique suitable for completing multistage tasks that required several shape changes.
“We have a stronger correlation between action points that are closer to each other, and I think that is key to making this work so well,” says Chen.
While it may be many years before shape-shifting robots are deployed in the real world, Chen and his collaborators hope their work inspires other scientists not only to study reconfigurable soft robots but also to think about leveraging 2D action spaces for other complex control problems.
The Computer Science and Artificial Intelligence Laboratory (CSAIL) is a research institute at the Massachusetts Institute of Technology (MIT) formed by the 2003 merger of the Laboratory for Computer Science (LCS) and the Artificial Intelligence Laboratory (AI Lab). Housed within the Ray and Maria Stata Center, CSAIL is the largest on-campus laboratory as measured by research scope and membership. It is part of the Schwarzman College of Computing but is also overseen by the MIT Vice President of Research.
Research activities
CSAIL’s research activities are organized around a number of semi-autonomous research groups, each of which is headed by one or more professors or research scientists. These groups are divided up into seven general areas of research:
Artificial intelligence
Computational biology
Graphics and vision
Language and learning
Theory of computation
Robotics
Systems (includes computer architecture, databases, distributed systems, networks and networked systems, operating systems, programming methodology, and software engineering among others)
In addition, CSAIL hosts the World Wide Web Consortium (W3C).
The MIT Stephen A. Schwarzman College of Computing is a college at the Massachusetts Institute of Technology. Announced in 2018 to address the growing applications of computing technology, the college is an Institute-wide academic unit that works alongside MIT’s five Schools of Architecture and Planning, Engineering, Humanities, Arts, and Social Sciences, Science, and Management. The college emphasizes artificial intelligence research, interdisciplinary applications of computing, and social and ethical responsibilities of computing. It aims to be an interdisciplinary hub for work in artificial intelligence, computer science, data science, and related fields. Its creation was the first significant change to MIT’s academic structure since the early 1950s.
The MIT Schwarzman College of Computing is named after The Blackstone Group chairman Stephen A. Schwarzman, who donated $350 million of the college’s $1.1 billion funding commitment. The college’s funding sources were met with criticism, with students and staff contrasting MIT’s stated emphasis on ethics against Schwarzman’s controversial business practices and support for Donald Trump.
Academics and research
The Schwarzman College of Computing has one academic department and several research enterprises which also have degree programs:
Department of Electrical Engineering and Computer Science (EECS, more commonly known at MIT as Course 6), which is jointly administered with the School of Engineering. Upon creation of the college, the department formerly only in the School of Engineering was reorganized into three “overlapping subunits”:
Electrical Engineering (EE)
Computer Science (CS)
Artificial Intelligence and Decision-Making (AI+D)
Operations Research Center (ORC), jointly administered with the MIT Sloan School of Management
Institute for Data, Systems and Society (IDSS)
Technology and Policy Program (TPP, adegree program)
Sociotechnical Systems Research Center (SSRC)
Center for Computational Science and Engineering (CCSE, renamed from Center for Computational Engineering upon formation of the college)
The non-degree-granting research labs which are part of the college are:
MIT Computer Science and Artificial Intelligence Laboratory (CSAIL)
MIT Laboratory for Information and Decision Systems (LIDS)
Quest for Intelligence
MIT-IBM Watson AI Lab
MIT Abdul Latif Jameel Clinic for Machine Learning in Health
The establishment of the college added 50 new faculty positions to the university. Half of these positions focus on computer science, while the other half are jointly appointed in collaboration with other departments in the Architecture and Planning, Engineering, Humanities, Arts, and Social Sciences, Science, and Management. The New York Times described the college’s structure as an effort to “alter traditional academic thinking and practice” and allow the university to more effectively bring computing to other fields.
The creation of the College of Computing also started the development of three additional programs meant to integrate closely with other MIT computing activities, for which plans have not been finalized:
Social and Ethical Responsibilities of Computing (SERC) aims to develop “responsible habits of mind and action” regarding computing technology. SERC facilitates the teaching of ethics throughout MIT courses, conducts research in social, ethical, and policy implications of technology, and coordinates public forums regarding technology and public policy. Common Ground for Computing Education coordinates interdepartmental teaching in computing, supporting interdisciplinary courses, majors, and minors on computing and its applications. Center for Advanced Studies of Computing hosts research fellows and assists project-oriented programs in computing-related topics.
EECS brings the world’s most brilliant faculty and students together to innovate and explore covering the full range of computer, information and energy systems. From foundational hardware and software systems, to cutting-edge machine learning models and computational methods to address critical societal problems, our work changes the world.
The MIT School of Engineering is one of the five schools of the Massachusetts Institute of Technology, located in Cambridge, Massachusetts. The School of Engineering has eight academic departments and two interdisciplinary institutes. The School grants SB, MEng, SM, engineer’s degrees, and PhD or ScD degrees. The school is the largest at MIT as measured by undergraduate and graduate enrollments and faculty members.
Departments and initiatives:
Departments:
Aeronautics and Astronautics (Course 16)
Biological Engineering (Course 20)
Chemical Engineering (Course 10)
Civil and Environmental Engineering (Course 1)
Electrical Engineering and Computer Science (Course 6, joint department with MIT Schwarzman College of Computing)
Materials Science and Engineering (Course 3)
Mechanical Engineering (Course 2)
Nuclear Science and Engineering (Course 22)
Institutes:
Institute for Medical Engineering and Science
Health Sciences and Technology program (joint MIT-Harvard, “HST” in the course catalog)
(Departments and degree programs are commonly referred to by course catalog numbers on campus.)
Laboratories and research centers
Abdul Latif Jameel Water and Food Systems Lab
Center for Advanced Nuclear Energy Systems
Center for Computational Engineering
Center for Materials Science and Engineering
Center for Ocean Engineering
Center for Transportation and Logistics
Industrial Performance Center
Institute for Soldier Nanotechnologies
Koch Institute for Integrative Cancer Research
Laboratory for Information and Decision Systems
Laboratory for Manufacturing and Productivity
Materials Processing Center
Microsystems Technology Laboratories
MIT Lincoln Laboratory Beaver Works Center
Novartis-MIT Center for Continuous Manufacturing
Ocean Engineering Design Laboratory
Research Laboratory of Electronics
SMART Center
Sociotechnical Systems Research Center
Tata Center for Technology and Design
USPS “Forever” postage stamps celebrating Innovation at MIT.
Founded in 1861 in response to the increasing industrialization of the United States, Massachusetts Institute of Technology adopted a European polytechnic university model and stressed laboratory instruction in applied science and engineering. It has since played a key role in the development of many aspects of modern science, engineering, mathematics, and technology, and is widely known for its innovation and academic strength. It is frequently regarded as one of the most prestigious universities in the world.
Nobel laureates, Turing Award winners, and Fields Medalists have been affiliated with MIT as alumni, faculty members, or researchers. In addition, National Medal of Science recipients, National Medals of Technology and Innovation recipients, MacArthur Fellows, Marshall Scholars, Mitchell Scholars, Schwarzman Scholars, astronauts, and Chief Scientists of the U.S. Air Force have been affiliated with The Massachusetts Institute of Technology. The university also has a strong entrepreneurial culture and MIT alumni have founded or co-founded many notable companies. The Massachusetts Institute of Technology is a member of the Association of American Universities.
Foundation and vision
In 1859, a proposal was submitted to the Massachusetts General Court to use newly filled lands in Back Bay, Boston for a “Conservatory of Art and Science”, but the proposal failed. A charter for the incorporation of the Massachusetts Institute of Technology, proposed by William Barton Rogers, was signed by John Albion Andrew, the governor of Massachusetts, on April 10, 1861.
Rogers, a professor from The University of Virginia , wanted to establish an institution to address rapid scientific and technological advances. He did not wish to found a professional school, but a combination with elements of both professional and liberal education, proposing that:
“The true and only practicable object of a polytechnic school is, as I conceive, the teaching, not of the minute details and manipulations of the arts, which can be done only in the workshop, but the inculcation of those scientific principles which form the basis and explanation of them, and along with this, a full and methodical review of all their leading processes and operations in connection with physical laws.”
The Rogers Plan reflected the German research university model, emphasizing an independent faculty engaged in research, as well as instruction oriented around seminars and laboratories.
Early developments
Two days after The Massachusetts Institute of Technology was chartered, the first battle of the Civil War broke out. After a long delay through the war years, MIT’s first classes were held in the Mercantile Building in Boston in 1865. The new institute was founded as part of the Morrill Land-Grant Colleges Act to fund institutions “to promote the liberal and practical education of the industrial classes” and was a land-grant school. In 1863 under the same act, the Commonwealth of Massachusetts founded the Massachusetts Agricultural College, which developed as The University of Massachusetts- Amherst . In 1866, the proceeds from land sales went toward new buildings in the Back Bay.
The Massachusetts Institute of Technology was informally called “Boston Tech”. The institute adopted the European polytechnic university model and emphasized laboratory instruction from an early date. Despite chronic financial problems, the institute saw growth in the last two decades of the 19th century under President Francis Amasa Walker. Programs in electrical, chemical, marine, and sanitary engineering were introduced, new buildings were built, and the size of the student body increased to more than one thousand.
The curriculum drifted to a vocational emphasis, with less focus on theoretical science. The fledgling school still suffered from chronic financial shortages which diverted the attention of the MIT leadership. During these “Boston Tech” years, The Massachusetts Institute of Technology faculty and alumni rebuffed Harvard University president (and former MIT faculty) Charles W. Eliot’s repeated attempts to merge MIT with Harvard College’s Lawrence Scientific School. There would be at least six attempts to absorb MIT into Harvard. In its cramped Back Bay location, MIT could not afford to expand its overcrowded facilities, driving a desperate search for a new campus and funding. Eventually, the MIT Corporation approved a formal agreement to merge with Harvard, over the vehement objections of MIT faculty, students, and alumni. However, a 1917 decision by the Massachusetts Supreme Judicial Court effectively put an end to the merger scheme.
In 1916, The Massachusetts Institute of Technology administration and the MIT charter crossed the Charles River on the ceremonial barge Bucentaur built for the occasion, to signify MIT’s move to a spacious new campus largely consisting of filled land on a one-mile-long (1.6 km) tract along the Cambridge side of the Charles River. The neoclassical “New Technology” campus was designed by William W. Bosworth and had been funded largely by anonymous donations from a mysterious “Mr. Smith”, starting in 1912. In January 1920, the donor was revealed to be the industrialist George Eastman of Rochester, New York, who had invented methods of film production and processing, and founded Eastman Kodak. Between 1912 and 1920, Eastman donated $20 million ($236.6 million in 2015 dollars) in cash and Kodak stock to MIT.
Curricular reforms
In the 1930s, President Karl Taylor Compton and Vice-President (effectively Provost) Vannevar Bush emphasized the importance of pure sciences like physics and chemistry and reduced the vocational practice required in shops and drafting studios. The Compton reforms “renewed confidence in the ability of the Institute to develop leadership in science as well as in engineering”. Unlike Ivy League schools, The Massachusetts Institute of Technology catered more to middle-class families, and depended more on tuition than on endowments or grants for its funding. The school was elected to the Association of American Universities in 1934.
Still, as late as 1949, the Lewis Committee lamented in its report on the state of education at The Massachusetts Institute of Technology that “the Institute is widely conceived as basically a vocational school”, a “partly unjustified” perception the committee sought to change. The report comprehensively reviewed the undergraduate curriculum, recommended offering a broader education, and warned against letting engineering and government-sponsored research detract from the sciences and humanities. The School of Humanities, Arts, and Social Sciences and the MIT Sloan School of Management were formed in 1950 to compete with the powerful Schools of Science and Engineering. Previously marginalized faculties in the areas of economics, management, political science, and linguistics emerged into cohesive and assertive departments by attracting respected professors and launching competitive graduate programs. The School of Humanities, Arts, and Social Sciences continued to develop under the successive terms of the more humanistically oriented presidents Howard W. Johnson and Jerome Wiesner between 1966 and 1980.
The Massachusetts Institute of Technology‘s involvement in military science surged during World War II. In 1941, Vannevar Bush was appointed head of the federal Office of Scientific Research and Development and directed funding to only a select group of universities, including MIT. Engineers and scientists from across the country gathered at The Massachusetts Institute of Technology’s Radiation Laboratory [now The Lincoln Laboratory], established in 1940 to assist the British military in developing microwave radar. The work done there significantly affected both the war and subsequent research in the area. Other defense projects included gyroscope-based and other complex control systems for gunsight, bombsight, and inertial navigation under Charles Stark Draper’s Instrumentation Laboratory; the development of a digital computer for flight simulations under Project Whirlwind; and high-speed and high-altitude photography under Harold Edgerton. By the end of the war, The Massachusetts Institute of Technology became the nation’s largest wartime R&D contractor (attracting some criticism of Bush), employing nearly 4000 in the Radiation Laboratory alone and receiving in excess of $100 million ($1.2 billion in 2015 dollars) before 1946. Work on defense projects continued even after then. Post-war government-sponsored research at MIT included SAGE and guidance systems for ballistic missiles and Project Apollo.
These activities affected The Massachusetts Institute of Technology profoundly. A 1949 report noted the lack of “any great slackening in the pace of life at the Institute” to match the return to peacetime, remembering the “academic tranquility of the prewar years”, though acknowledging the significant contributions of military research to the increased emphasis on graduate education and rapid growth of personnel and facilities. The faculty doubled and the graduate student body quintupled during the terms of Karl Taylor Compton, president of The Massachusetts Institute of Technology between 1930 and 1948; James Rhyne Killian, president from 1948 to 1957; and Julius Adams Stratton, chancellor from 1952 to 1957, whose institution-building strategies shaped the expanding university. By the 1950s, The Massachusetts Institute of Technology no longer simply benefited the industries with which it had worked for three decades, and it had developed closer working relationships with new patrons, philanthropic foundations and the federal government.
In late 1960s and early 1970s, student and faculty activists protested against the Vietnam War and The Massachusetts Institute of Technology ‘s defense research. In this period The Massachusetts Institute of Technology’s various departments were researching helicopters, smart bombs and counterinsurgency techniques for the war in Vietnam as well as guidance systems for nuclear missiles. The Union of Concerned Scientists was founded on March 4, 1969 during a meeting of faculty members and students seeking to shift the emphasis on military research toward environmental and social problems. The Massachusetts Institute of Technology ultimately divested itself from the Instrumentation Laboratory and moved all classified research off-campus to The MIT Lincoln Laboratory facility in 1973 in response to the protests. The student body, faculty, and administration remained comparatively unpolarized during what was a tumultuous time for many other universities. Johnson was seen to be highly successful in leading his institution to “greater strength and unity” after these times of turmoil. However, six Massachusetts Institute of Technology students were sentenced to prison terms at this time and some former student leaders, such as Michael Albert and George Katsiaficas, are still indignant about MIT’s role in military research and its suppression of these protests. (Richard Leacock’s film, November Actions, records some of these tumultuous events.)
In the 1980s, there was more controversy at The Massachusetts Institute of Technology over its involvement in SDI (space weaponry) and CBW (chemical and biological warfare) research. More recently, The Massachusetts Institute of Technology’s research for the military has included work on robots, drones and ‘battle suits’.
Recent history
The Massachusetts Institute of Technology has kept pace with and helped to advance the digital age. In addition to developing the predecessors to modern computing and networking technologies, students, staff, and faculty members at Project MAC, the Artificial Intelligence Laboratory [now The MIT Computer Science and Artificial Intelligence Laboratory], and the Tech Model Railroad Club wrote some of the earliest interactive computer video games like Spacewar! and created much of modern hacker slang and culture. Several major computer-related organizations have originated at MIT since the 1980s: Richard Stallman’s GNU Project and the subsequent Free Software Foundation were founded in the mid-1980s at the AI Lab; The MIT Media Lab was founded in 1985 by Nicholas Negroponte and Jerome Wiesner to promote research into novel uses of computer technology; The World Wide Web Consortium standards organization was founded at The Laboratory for Computer Science in 1994 by Tim Berners-Lee; The MIT OpenCourseWare project has made course materials for over 2,000 Massachusetts Institute of Technology classes available online free of charge since 2002; and the One Laptop per Child initiative to expand computer education and connectivity to children worldwide was launched in 2005.
The Massachusetts Institute of Technology was named a sea-grant college in 1976 to support its programs in oceanography and marine sciences and was named a space-grant college in 1989 to support its aeronautics and astronautics programs. Despite diminishing government financial support over the past quarter century, MIT launched several successful development campaigns to significantly expand the campus: new dormitories and athletics buildings on west campus; The Tang Center for Management Education; several buildings in the northeast corner of campus supporting research into biology, brain and cognitive sciences, genomics, biotechnology, and cancer research; and a number of new “backlot” buildings on Vassar Street including the Stata Center. Construction on campus in the 2000s included expansions of the Media Lab, the Sloan School’s eastern campus, and graduate residences in the northwest. In 2006, President Hockfield launched The MIT Energy Research Council [now The MIT Energy Initiative] to investigate the interdisciplinary challenges posed by increasing global energy consumption.
In 2001, inspired by the open source and open access movements, The Massachusetts Institute of Technology launched “OpenCourseWare” to make the lecture notes, problem sets, syllabi, exams, and lectures from the great majority of its courses available online for no charge, though without any formal accreditation for coursework completed. While the cost of supporting and hosting the project is high, OCW expanded in 2005 to include other universities as a part of The OpenCourseWare Consortium, which currently includes more than 250 academic institutions with content available in at least six languages. In 2011, The Massachusetts Institute of Technology announced it would offer formal certification (but not credits or degrees) to online participants completing coursework in its “MITx” program, for a modest fee. The “edX” online platform supporting MITx was initially developed in partnership with Harvard and its analogous “Harvardx” initiative. The courseware platform is open source, and other universities have already joined and added their own course content. In March 2009 The Massachusetts Institute of Technology faculty adopted an open-access policy to make its scholarship publicly accessible online.
The Massachusetts Institute of Technology has its own police force. Three days after the Boston Marathon bombing of April 2013, MIT Police patrol officer Sean Collier was fatally shot by the suspects Dzhokhar and Tamerlan Tsarnaev, setting off a violent manhunt that shut down the campus and much of the Boston metropolitan area for a day. One week later, Collier’s memorial service was attended by more than 10,000 people, in a ceremony hosted by The Massachusetts Institute of Technology community with thousands of police officers from the New England region and Canada. On November 25, 2013, The Massachusetts Institute of Technology announced the creation of the Collier Medal, to be awarded annually to “an individual or group that embodies the character and qualities that Officer Collier exhibited as a member of The Massachusetts Institute of Technology community and in all aspects of his life”. The announcement further stated that “Future recipients of the award will include those whose contributions exceed the boundaries of their profession, those who have contributed to building bridges across the community, and those who consistently and selflessly perform acts of kindness”.
In September 2017, the school announced the creation of an artificial intelligence research lab called The MIT-IBM Watson AI Lab. IBM will spend $240 million over the next decade, and the lab will be staffed by MIT and IBM scientists. In October 2018 MIT announced that it would open the new Schwarzman College of Computing dedicated to the study of artificial intelligence, named after lead donor and The Blackstone Group CEO Stephen Schwarzman. The focus of the new college is to study not just AI, but interdisciplinary AI education, and how AI can be used in fields as diverse as history and biology. The cost of buildings and new faculty for the new college is expected to be $1 billion upon completion.
It was designed to open the field of gravitational-wave astronomy through the detection of gravitational waves predicted by general relativity. Gravitational waves were detected for the first time by the LIGO detector in 2015. For contributions to the LIGO detector and the observation of gravitational waves, two Caltech physicists, Kip Thorne and Barry Barish, and Massachusetts Institute of Technology physicist Rainer Weiss won the Nobel Prize in physics in 2017. Weiss, who is also a Massachusetts Institute of Technology graduate, designed the laser interferometric technique, which served as the essential blueprint for the LIGO.
The mission of The Massachusetts Institute of Technology is to advance knowledge and educate students in science, technology, and other areas of scholarship that will best serve the nation and the world in the twenty-first century. We seek to develop in each member of The Massachusetts Institute of Technology community the ability and passion to work wisely, creatively, and effectively for the betterment of humankind.